Midterm
- Six questions
- Thursday (10:00AM) to Tuesday (10:00AM)
Pronghorn
Connect to Pronghorn
ssh <yourID>@pronghorn.rc.unr.edu
What is file permission?
- (rw-) (rw-) (r--) 1 john sap
| | | |
type owner group others
How to download file from Pronghorn in your local
scp <yourID>@pronghorn.rc.unr.edu:~/[FOLDER AND FILE] [LOCAL directory]
Conda
Conda installation
search on web browser [software name & conda] on Google ex: 'hisat2 conda'
conda install [package name]
Conda export
export your environment to rnaseq.yaml
conda env export > [Name].yaml
Sequencing
Illumina sequencing
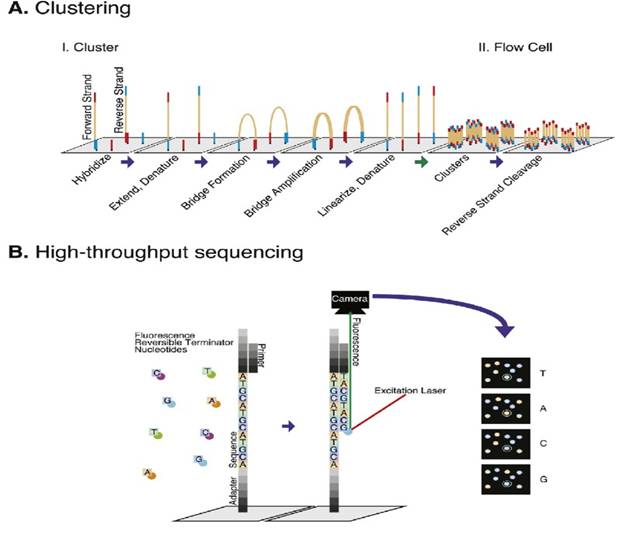
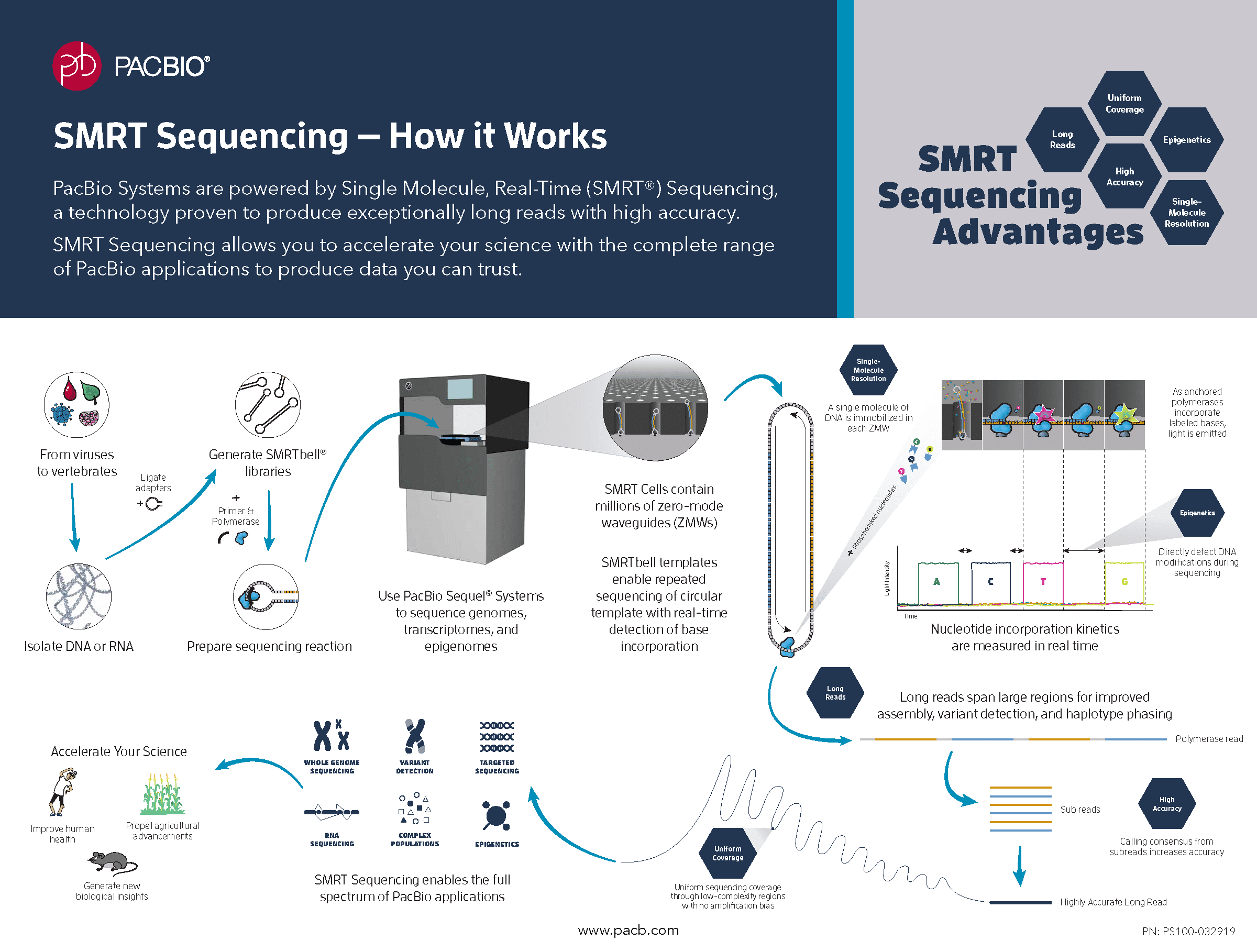
PacBio sequencing
File format
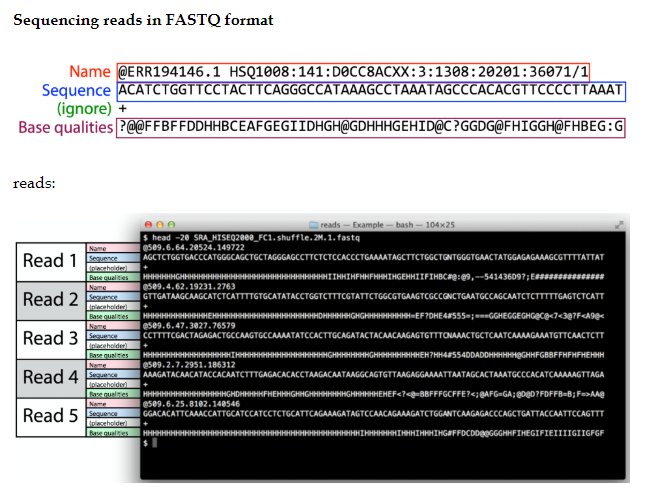
Fasta file

Fastq file

GFF format
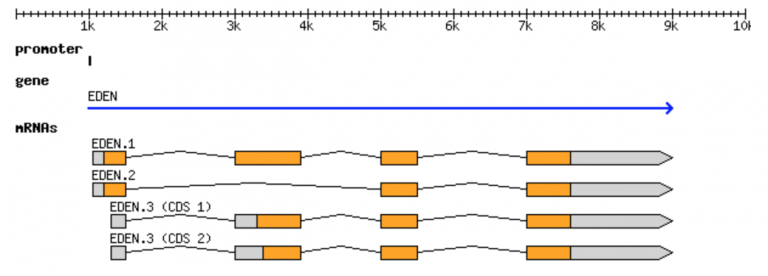
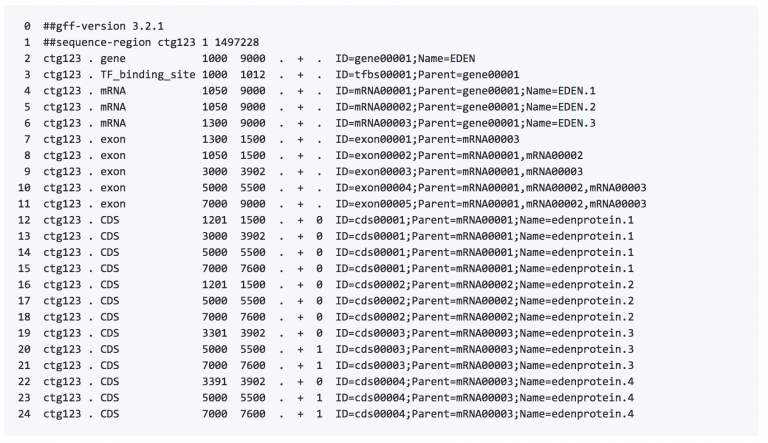
- Sequence ID
- Source
- Describes the algorithm or the procedure that generated this feature. Typically Genescane or Genebank, respectively.
- Feature Type
- Describes what the feature is (mRNA, domain, exon, etc.). These terms are constrained to the Sequence Ontology terms.
- Feature Start
- Feature End
- Score
- Typically E-values for sequence similarity and P-values for predictions.
- Strand
- Phase
- Indicates where the feature begins with reference to the reading frame. The phase is one of the integers 0, 1, or 2, indicating the number of bases that should be removed from the beginning of this feature to reach the first base of the next codon. 9 .Atributes
- A semicolon-separated list of tag-value pairs, providing additional information about each feature. Some of these tags are predefined, e.g. ID, Name, Alias, Parent . You can see the full list here.
RNA Sequencing

- The transcriptome is spatially and temporally dynamic
- Data comes from functional units (coding regions)
- Only a tiny fraction of the genome
Paper reading
Please read this paper
https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-0881-8
Conda
- Dependencies is one of the main reasons to use Conda.
Sometimes, install a package is not as straight forward as you think. Imagine a case like this: You want to install package Matplotlib, when installing, it asks you to install Numpy, and Scipy, because Matplotlib need these Numpy and Scipy to work. They are called the dependencies of Matplotlib. For Numpy and Scipy, they may have their own dependencies. These require even more packages.
Conda env clean
conda clean --all
Conda create enviroment
conda create -n review python=3
Conda activate enviroment
conda activate review
Install software
conda install -c bioconda -c anaconda trinity samtools multiqc fastqc rsem jellyfish bowtie2 salmon trim-galore fastqc bioconductor-ctc bioconductor-deseq2 bioconductor-edger bioconductor-biobase bioconductor-qvalue r-ape r-gplots r-fastcluster
conda install -c anaconda openblas
conda install nano
conda install -c eumetsat tree
conda install -c lmfaber transrate
Check installation
conda list
Basic Unix/Linux command
cd
cd /data/gpfs/assoc/bch709-4/<YOUR_ID>
mkdir
mkdir BCH709_midterm
cd BCH709_midterm
pwd
pwd
wget
file download
wget https://www.dropbox.com/s/yqvfm70yz79jvij/fasta.zip https://www.dropbox.com/s/jjz6aip3euh0d7q/fastq.tar
wget https://www.dropbox.com/s/szzyb3l4243xcsu/bch709.py
Decompress tar file
tar xvf fastq.tar
ls
Decompress zip file
unzip fasta.zip
ls
gz file
zcat
pipe
wc
rm
RNA-Seq

Advanced bioinformatics tools
Seqkit
https://plantgenomicslab.github.io/BCH709/seqkit_tutorial/index.html
Trim-Galore
Install Trim Galore
conda install -c bioconda -c conda-forge trim-galore
Run trimming
trim_galore --help
trim_galore --paired --three_prime_clip_R1 20 --three_prime_clip_R2 20 --cores 2 --max_n 40 --gzip -o trim pair1.fastq.gz pair2.fastq.gz --fastqc
HISAT2
Example
Install HISAT2 (graph FM index, spin off version Burrows-Wheeler Transform)
conda install -c conda-forge -c bioconda hisat2
Download reference sequence
wget https://nevada.box.com/shared/static/5v14j6gjt16c7k5d42b7g51csjmmj16l.fasta -O bch709.fasta
HISAT2 indexing
hisat2-build --help
hisat2-build bch709.fasta bch709
HISAT2 mapping
hisat2 -x bch709 --threads <YOUR CPU COUNT> -1 trim/paired1_val_1.fq.gz -2 trim/paired2_val_2.fq.gz -S align.sam 2> summarymetrics.txt
MultiQC
multiqc .
SAMtools
SAM (Sequence Alignment/Map) format is a generic format for storing large nucleotide sequence alignments. SAM aims to be a format that:
- Is flexible enough to store all the alignment information generated by various alignment programs;
- Is simple enough to be easily generated by alignment programs or converted from existing alignment formats;
- Is compact in file size;
- Allows most of operations on the alignment to work on a stream without loading the whole alignment into memory;
- Allows the file to be indexed by genomic position to efficiently retrieve all reads aligning to a locus.
SAM Tools provide various utilities for manipulating alignments in the SAM format, including sorting, merging, indexing and generating alignments in a per-position format. http://samtools.sourceforge.net/
samtools view -Sb align.sam -o align.bam
samtools sort align.bam -o align_sort.bam
samtools index align_sort.bam
Quantification • Counts
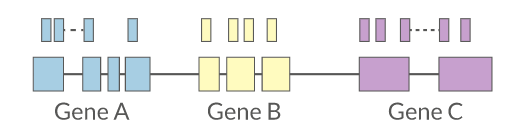
- Read counts = gene expression
- Reads can be quantified on any feature (gene, transcript, exon etc)
- Intersection on gene models
- Gene/Transcript level

featureCounts HTSeq RSEM Cufflinks Rcount
conda install -c conda-forge -c bioconda subread
wget https://www.dropbox.com/s/e9dvdkrl9dta4qg/bch709.gtf
featureCounts -p -a bch709.gtf align_sort.bam -o counts.txt
References:
- Conda documentation https://docs.conda.io/en/latest/
- Conda-forge https://conda-forge.github.io/
- BioConda https://bioconda.github.io/