HOME WORK
- de Bruijn graph construction (10 pts)
- Draw (by hand) the de Bruijn graph for the following reads using k=3 (assume all reads are from the forward strand, no sequencing errors) AGT
ATG
CAT
GTA
GTT
TAC
TAG
TGT
TTA
HOME WORK ANSWER
HPC and Cloud
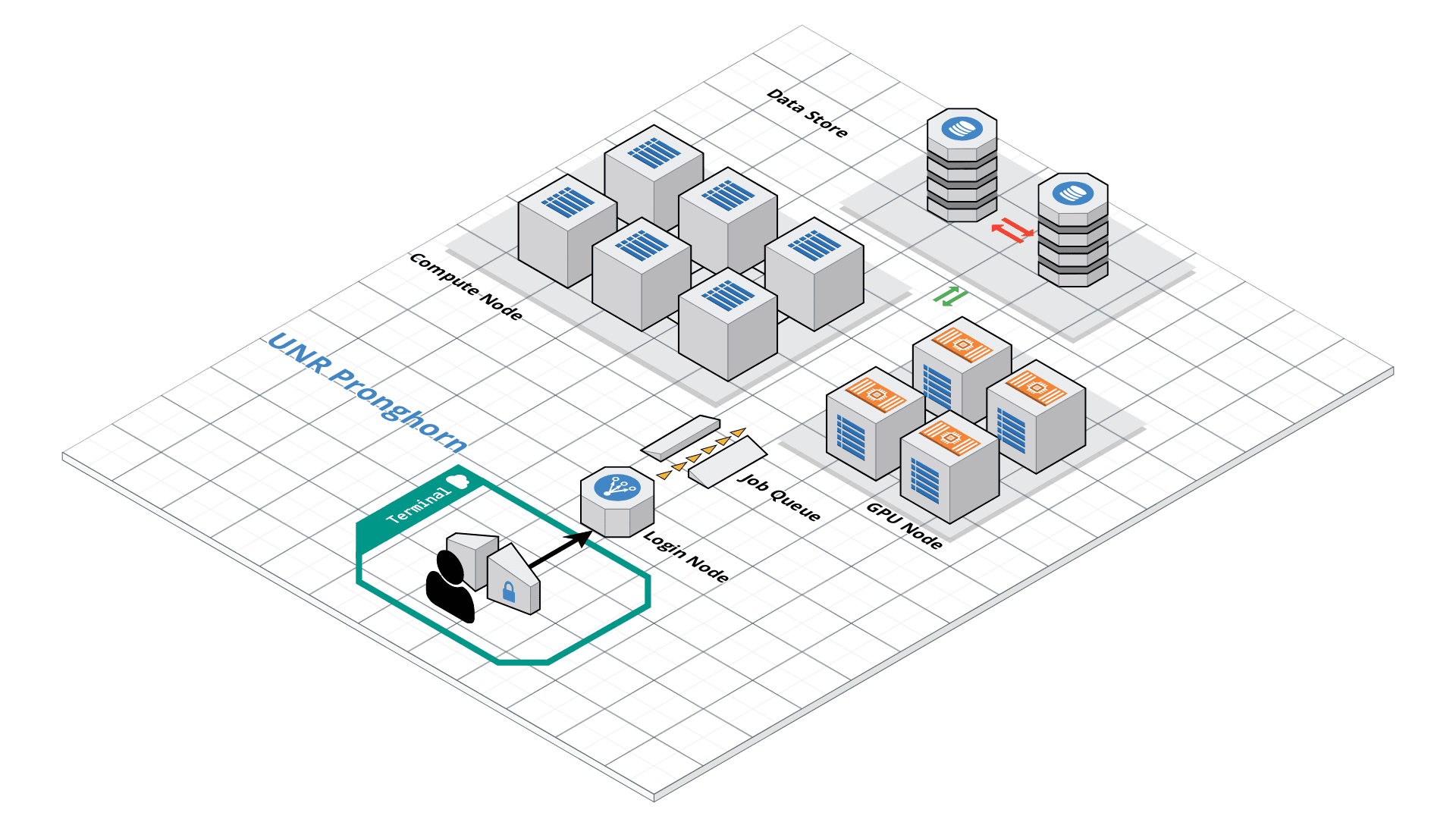
This exercise mainly deals with using HPC clusters for large scale data (Next Generation Sequencing analysis, Genome annotation, evolutionary studies etc.). These clusters have several processors with large amounts of RAM (compared to typical desktop/laptop), which makes it ideal for running programs that are computationally intensive. The operating system of these clusters are primarily UNIX and are mainly operated via command line. All the commands that you have learned in the previous exercises can be used on HPC.
SSH
The ssh command is pre-installed. It means Secure Shell.
ssh <YOURID>@pronghorn.rc.unr.edu

Bash on Pronghorn
When you work with the command line and the bash shell frequently, you will want to customize the environment. This can mean changing environment variables, such as where the shell looks for commands or how the prompt looks, or adding customized commands.
nano ~/.bashrc
###setting history length see HISTSIZE and HISTFILESIZE in bash(1)
HISTSIZE=1000
HISTFILESIZE=3000
###Alias
alias vi='vim'
alias grep='grep --color=auto'
alias egrep='egrep --color=auto'
alias ls='ls --color=auto'
alias ll='ls -alhg'
alias la='ls -A'
alias l='ls -CF'
##Shell color
tty -s && export PS1="\[\033[38;5;5m\]\u\[$(tput sgr0)\]\[\033[38;5;15m\] @ \[$(tput bold)\]\[$(tput sgr0)\]\[\033[38;5;1m\]\H\[$(tput sgr0)\]\[$(tput sgr0)\]\[\033[38;5;15m\] \[$(tput sgr0)\]\[\033[38;5;178m\]\T\[$(tput sgr0)\]\[\033[38;5;2m\]\w\[$(tput sgr0)\]\[\033[38;5;15m\] \n\[$(tput sgr0)\]"
##Module
module load slurm
module load hdf5_18 gcc/5.4.0 blas/gcc/64/3.8.0
module load singularity
source ~/.bashrc
File transfer
- There are a number of ways to transfer data to and from HPC clusters. Which you should use depends on several factors, including the ease of use for you personally, connection speed and bandwidth, and the size and number of files which you intend to transfer. Most common options include scp, rsync (command line) and SCP and SFTP clients (GUI). scp (secure copy) is a simple way of transferring files between two machines that use the SSH (Secure SHell) protocol. You may use scp to connect to any system where you have SSH (login) access. scp is available as a protocol choice in some graphical file transfer programs and also as a command line program on most Linux, UNIX, and Mac OS X systems. scp can copy single files, but will also recursively copy directory contents if given a directory name. scp can be used as follows:
scp sourcefile username@pronghorn.rc.unr.edu:somedirectory/(to a remote system from local)
scp username@pronghorn.rc.unr.edu:somedirectory/sourcefile destinationfile(from a remote system to local)
scp -r SourceDirectory/ username@pronghorn.rc.unr.edu:somedirectory/(recursive directory copy to a remote system from local)
- rsync is a fast and extraordinarily versatile file copying tool. It can synchronize file trees across local disks, directories or across a network
rsync -avhP path/to/SourceDirectory username@pronghorn.rc.unr.edu:somedirectory/(Synchronize a local directory with the remote server directory)
rsync -avhP username@pronghorn.rc.unr.edu:SourceDirectory/ path/to/Destination/(Synchronize a remote directory with the local directory)
Slurm Quick Start Tutorial
Resource sharing on a supercomputer dedicated to technical and/or scientific computing is often organized by a piece of software called a resource manager or job scheduler. Users submit jobs, which are scheduled and allocated resources (CPU time, memory, etc.) by the resource manager.
Slurm is a resource manager and job scheduler designed to do just that, and much more. It was originally created by people at the Livermore Computing Center, and has grown into a full-fledge open-source software backed up by a large community, commercially supported by the original developers, and installed in many of the Top500 supercomputers.
Gathering information Slurm offers many commands you can use to interact with the system. For instance, the sinfo command gives an overview of the resources offered by the cluster, while the squeue command shows to which jobs those resources are currently allocated.
By default, sinfo lists the partitions that are available. A partition is a set of compute nodes (computers dedicated to… computing) grouped logically. Typical examples include partitions dedicated to batch processing, debugging, post processing, or visualization.
sinfo
Show the State of Nodes
sinfo --all
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
cpu-s2-core-0 up 14-00:00:0 2 mix cpu-[8-9]
cpu-s2-core-0 up 14-00:00:0 7 alloc cpu-[1-2,4-6,78-79]
cpu-s2-core-0 up 14-00:00:0 44 idle cpu-[0,3,7,10-47,64,76-77]
cpu-s3-core-0* up 2:00:00 2 mix cpu-[8-9]
cpu-s3-core-0* up 2:00:00 7 alloc cpu-[1-2,4-6,78-79]
cpu-s3-core-0* up 2:00:00 44 idle cpu-[0,3,7,10-47,64,76-77]
gpu-s2-core-0 up 14-00:00:0 11 idle gpu-[0-10]
cpu-s6-test-0 up 15:00 2 idle cpu-[65-66]
cpu-s1-pgl-0 up 14-00:00:0 1 mix cpu-49
cpu-s1-pgl-0 up 14-00:00:0 1 alloc cpu-48
cpu-s1-pgl-0 up 14-00:00:0 2 idle cpu-[50-51]
In the above example, we see two partitions, named batch and debug. The latter is the default partition as it is marked with an asterisk. All nodes of the debug partition are idle, while two of the batch partition are being used.
The sinfo command also lists the time limit (column TIMELIMIT) to which jobs are subject. On every cluster, jobs are limited to a maximum run time, to allow job rotation and let every user a chance to see their job being started. Generally, the larger the cluster, the smaller the maximum allowed time. You can find the details on the cluster page.
You can actually specify precisely what information you would like sinfo to output by using its –format argument. For more details, have a look at the command manpage with man sinfo.
sinfo advance
sinfo $OPTIONS -o "%13n %8t %4c %8z %15C %8O %8m %8G %18P %f" | egrep s2
squeue
The squeue command shows the list of jobs which are currently running (they are in the RUNNING state, noted as ‘R’) or waiting for resources (noted as ‘PD’, short for PENDING).
squeue
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
983204 cpu-s2-co neb_K jzhang23 R 6-09:05:47 1 cpu-6
983660 cpu-s2-co RT3.sl yinghanc R 12:56:17 1 cpu-9
983659 cpu-s2-co RT4.sl yinghanc R 12:56:21 1 cpu-8
983068 cpu-s2-co Gd-bound dcantu R 7-06:16:01 2 cpu-[78-79]
983067 cpu-s2-co Gd-unbou dcantu R 1-17:41:56 2 cpu-[1-2]
983472 cpu-s2-co ub-all dcantu R 3-10:05:01 2 cpu-[4-5]
982604 cpu-s1-pg wrap wyim R 12-14:35:23 1 cpu-49
983585 cpu-s1-pg wrap wyim R 1-06:28:29 1 cpu-48
983628 cpu-s1-pg wrap wyim R 13:44:46 1 cpu-49
SBATCH
Now the question is: How do you create a job?
A job consists in two parts: resource requests and job steps. Resource requests consist in a number of CPUs, computing expected duration, amounts of RAM or disk space, etc. Job steps describe tasks that must be done, software which must be run.
The typical way of creating a job is to write a submission script. A submission script is a shell script, e.g. a Bash script, whose comments, if they are prefixed with SBATCH, are understood by Slurm as parameters describing resource requests and other submissions options. You can get the complete list of parameters from the sbatch manpage man sbatch.
Important
The SBATCH directives must appear at the top of the submission file, before any other line except for the very first line which should be the shebang (e.g. #!/bin/bash). The script itself is a job step. Other job steps are created with the srun command. For instance, the following script, hypothetically named submit.sh,
nano submit.sh
#!/bin/bash
#SBATCH --job-name=test
#SBATCH --cpus-per-task=16
#SBATCH --time=10:00
#SBATCH --mem=1g
#SBATCH --mail-type=begin
#SBATCH --mail-type=end
#SBATCH --mail-user=<YOUR ID>@unr.edu
echo "Hello Pronghorn"
seq 1 80000
would request one CPU for 10 minutes, along with 1g of RAM, in the default queue. When started, the job would run a first job step srun hostname, which will launch the UNIX command hostname on the node on which the requested CPU was allocated. Then, a second job step will start the sleep command. Note that the –job-name parameter allows giving a meaningful name to the job and the –output parameter defines the file to which the output of the job must be sent.
Once the submission script is written properly, you need to submit it to slurm through the sbatch command, which, upon success, responds with the jobid attributed to the job. (The dollar sign below is the shell prompt)
$ chmod 775 submit.sh
$ sbatch submit.sh
sbatch: Submitted batch job 99999999
https://www.youtube.com/watch?v=U42qlYkzP9k&list=TLrtXVJajzvonT-8qcp5ZgtKCeyN3Pe4xv
Set up Conda
cd ~/
conda env remove --name rnaseq
wget https://www.dropbox.com/s/ed4b83m7z5ecy1y/bch709.yml
conda env create -n rnaseq -f bch709.yml
Running Trinity
Trinity is run via the script: ‘Trinity’ found in the base installation directory.
Usage info is as follows:
###############################################################################
#
# ______ ____ ____ ____ ____ ______ __ __
# | || \ | || \ | || || | |
# | || D ) | | | _ | | | | || | |
# |_| |_|| / | | | | | | | |_| |_|| ~ |
# | | | \ | | | | | | | | | |___, |
# | | | . \ | | | | | | | | | | |
# |__| |__|\_||____||__|__||____| |__| |____/
#
###############################################################################
#
# Required:
#
# --seqType <string> :type of reads: ( fa, or fq )
#
# --max_memory <string> :suggested max memory to use by Trinity where limiting can be enabled. (jellyfish, sorting, etc)
# provided in Gb of RAM, ie. '--max_memory 10G'
#
# If paired reads:
# --left <string> :left reads, one or more file names (separated by commas, not spaces)
# --right <string> :right reads, one or more file names (separated by commas, not spaces)
#
# Or, if unpaired reads:
# --single <string> :single reads, one or more file names, comma-delimited (note, if single file contains pairs, can use flag: --run_as_paired )
#
# Or,
# --samples_file <string> tab-delimited text file indicating biological replicate relationships.
# ex.
# cond_A cond_A_rep1 A_rep1_left.fq A_rep1_right.fq
# cond_A cond_A_rep2 A_rep2_left.fq A_rep2_right.fq
# cond_B cond_B_rep1 B_rep1_left.fq B_rep1_right.fq
# cond_B cond_B_rep2 B_rep2_left.fq B_rep2_right.fq
#
# # if single-end instead of paired-end, then leave the 4th column above empty.
#
####################################
## Misc: #########################
#
# --SS_lib_type <string> :Strand-specific RNA-Seq read orientation.
# if paired: RF or FR,
# if single: F or R. (dUTP method = RF)
# See web documentation.
#
# --CPU <int> :number of CPUs to use, default: 2
# --min_contig_length <int> :minimum assembled contig length to report
# (def=200)
#
# --long_reads <string> :fasta file containing error-corrected or circular consensus (CCS) pac bio reads
#
# --genome_guided_bam <string> :genome guided mode, provide path to coordinate-sorted bam file.
# (see genome-guided param section under --show_full_usage_info)
#
# --jaccard_clip :option, set if you have paired reads and
# you expect high gene density with UTR
# overlap (use FASTQ input file format
# for reads).
# (note: jaccard_clip is an expensive
# operation, so avoid using it unless
# necessary due to finding excessive fusion
# transcripts w/o it.)
#
# --trimmomatic :run Trimmomatic to quality trim reads
# see '--quality_trimming_params' under full usage info for tailored settings.
#
#
# --no_normalize_reads :Do *not* run in silico normalization of reads. Defaults to max. read coverage of 50.
# see '--normalize_max_read_cov' under full usage info for tailored settings.
# (note, as of Sept 21, 2016, normalization is on by default)
#
#
#
# --output <string> :name of directory for output (will be
# created if it doesn't already exist)
# default( your current working directory: "/Users/bhaas/GITHUB/trinityrnaseq/trinity_out_dir"
# note: must include 'trinity' in the name as a safety precaution! )
#
# --full_cleanup :only retain the Trinity fasta file, rename as ${output_dir}.Trinity.fasta
#
# --cite :show the Trinity literature citation
#
# --version :reports Trinity version (BLEEDING_EDGE) and exits.
#
# --show_full_usage_info :show the many many more options available for running Trinity (expert usage).
#
#
###############################################################################
#
# *Note, a typical Trinity command might be:
#
# Trinity --seqType fq --max_memory 50G --left reads_1.fq --right reads_2.fq --CPU 6
#
#
# and for Genome-guided Trinity:
#
# Trinity --genome_guided_bam rnaseq_alignments.csorted.bam --max_memory 50G
# --genome_guided_max_intron 10000 --CPU 6
#
# see: /Users/bhaas/GITHUB/trinityrnaseq/sample_data/test_Trinity_Assembly/
# for sample data and 'runMe.sh' for example Trinity execution
#
# For more details, visit: http://trinityrnaseq.github.io
#
###############################################################################
Trinity performs best with strand-specific data, in which case sense and antisense transcripts can be resolved. For protocols on strand-specific RNA-Seq, see: Borodina T, Adjaye J, Sultan M. A strand-specific library preparation protocol for RNA sequencing. Methods Enzymol. 2011;500:79-98. PubMed PMID: 21943893.
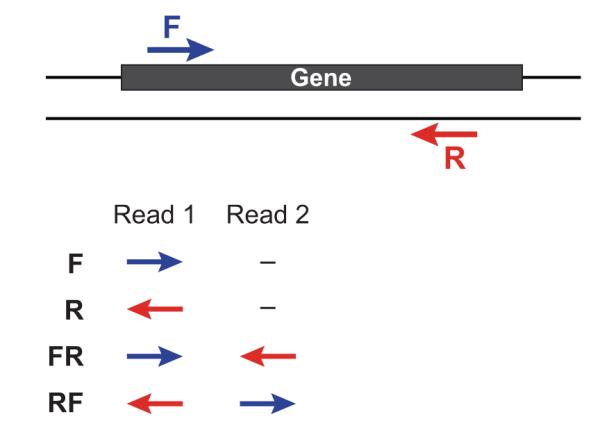
If you have strand-specific data, specify the library type. There are four library types:
- Paired reads:
- RF: first read (/1) of fragment pair is sequenced as anti-sense (reverse(R)), and second read (/2) is in the sense strand (forward(F)); typical of the dUTP/UDG sequencing method.
- FR: first read (/1) of fragment pair is sequenced as sense (forward), and second read (/2) is in the antisense strand (reverse)
- Unpaired (single) reads:
- F: the single read is in the sense (forward) orientation
- R: the single read is in the antisense (reverse) orientation
By setting the –SS_lib_type parameter to one of the above, you are indicating that the reads are strand-specific. By default, reads are treated as not strand-specific.
Other important considerations:
-
Whether you use Fastq or Fasta formatted input files, be sure to keep the reads oriented as they are reported by Illumina, if the data are strand-specific. This is because, Trinity will properly orient the sequences according to the specified library type. If the data are not strand-specific, no worries because the reads will be parsed in both orientations.
-
If you have both paired and unpaired data, and the data are NOT strand-specific, you can combine the unpaired data with the left reads of the paired fragments. Be sure that the unpaired reads have a /1 as a suffix to the accession value similarly to the left fragment reads. The right fragment reads should all have /2 as the accession suffix. Then, run Trinity using the –left and –right parameters as if all the data were paired.
-
If you have multiple paired-end library fragment sizes, set the ‘–group_pairs_distance’ according to the larger insert library. Pairings that exceed that distance will be treated as if they were unpaired by the Butterfly process.
-
by setting the ‘–CPU option’, you are indicating the maximum number of threads to be used by processes within Trinity. Note that Inchworm alone will be internally capped at 6 threads, since performance will not improve for this step beyond that setting)
Typical Trinity Command Line
A typical Trinity command for assembling non-strand-specific RNA-seq data would be like so, running the entire process on a single high-memory server (aim for ~1G RAM per ~1M ~76 base Illumina paired reads, but often much less memory is required):
Run Trinity like so:
Trinity --seqType fq --max_memory 50G \
--left reads_1.fq.gz --right reads_2.fq.gz --CPU 6
If you have multiple sets of fastq files, such as corresponding to multiple tissue types or conditions, etc., you can indicate them to Trinity like so:
Trinity --seqType fq --max_memory 50G \
--left condA_1.fq.gz,condB_1.fq.gz,condC_1.fq.gz \
--right condA_2.fq.gz,condB_2.fq.gz,condC_2.fq.gz \
--CPU 6
or better yet, create a ‘samples.txt’ file that describes the data like so:
# --samples_file <string> tab-delimited text file indicating biological replicate relationships.
# ex.
# cond_A cond_A_rep1 A_rep1_left.fq A_rep1_right.fq
# cond_A cond_A_rep2 A_rep2_left.fq A_rep2_right.fq
# cond_B cond_B_rep1 B_rep1_left.fq B_rep1_right.fq
# cond_B cond_B_rep2 B_rep2_left.fq B_rep2_right.fq
This same samples file can then be used later on with other downstream analysis steps, including expression quantification and differential expression analysis.
note that fastq files can be gzip-compressed as shown above, in which case they should require a ‘.gz’ extension.
File download
$ mkdir -p ~/rnaseq/transcriptome_assembly/fastq
$ cd ~/rnaseq/transcriptome_assembly/fastq
Download below files
https://www.dropbox.com/s/mzvempzve7uuwoa/KRWTD1_1.fastq.gz
https://www.dropbox.com/s/uv2a10w9wj9tcww/KRWTD1_2.fastq.gz
https://www.dropbox.com/s/ra0s10g2nag6axp/KRWTD2_1.fastq.gz
https://www.dropbox.com/s/jao5tb8a1hnzqw4/KRWTD2_2.fastq.gz
https://www.dropbox.com/s/4gwhz0t1d32cdnw/KRWTD3_1.fastq.gz
https://www.dropbox.com/s/wp2uk0nafdb74wp/KRWTD3_2.fastq.gz
https://www.dropbox.com/s/ctzo9k9n8qdpvio/KRWTW1_1.fastq.gz
https://www.dropbox.com/s/psiak4r2910sjsc/KRWTW1_2.fastq.gz
https://www.dropbox.com/s/so4zeuyqz64m80z/KRWTW2_1.fastq.gz
https://www.dropbox.com/s/2ggf2xdiydtehdw/KRWTW2_2.fastq.gz
https://www.dropbox.com/s/7bfgcq69cymb5yj/KRWTW3_1.fastq.gz
https://www.dropbox.com/s/lfif4qnbhnfes26/KRWTW3_2.fastq.gz
Run trimming
$ cd ~/rnaseq/transcriptome_assembly
$ pwd
$ nano trim.sh
#!/bin/bash
#SBATCH --job-name=<TRIM>
#SBATCH --cpus-per-task=16
#SBATCH --time=2:00:00
#SBATCH --mem-per-cpu=1g
#SBATCH --mail-type=begin
#SBATCH --mail-type=end
#SBATCH --mail-user=<YOUR ID>@unr.edu
fastqc ~/rnaseq/transcriptome_assembly/fastq/*.gz
trim_galore --paired --three_prime_clip_R1 20 --three_prime_clip_R2 20 --cores 16 --max_n 40 --gzip -o ~/rnaseq/transcriptome_assembly/trimmed_fastq ~/rnaseq/transcriptome_assembly/fastq/KRWTD1_1.fastq.gz ~/rnaseq/transcriptome_assembly/fastq/KRWTD1_2.fastq.gz
trim_galore --paired --three_prime_clip_R1 20 --three_prime_clip_R2 20 --cores 16 --max_n 40 --gzip -o ~/rnaseq/transcriptome_assembly/trimmed_fastq ~/rnaseq/transcriptome_assembly/fastq/KRWTD2_1.fastq.gz ~/rnaseq/transcriptome_assembly/fastq/KRWTD2_2.fastq.gz
.
.
.
fastqc ~/rnaseq/transcriptome_assembly/trimmed_fastq/*
Submit job
chmod 775 trim.sh
sbatch trim.sh
PLEASE CHECK YOUR MULTIQC
Merge gz file
zcat ~/rnaseq/transcriptome_assembly/trimmed_fastq/KRWTD1_1_val_1.fq.gz ........... >> merged_R1.fastq
zcat ~/rnaseq/transcriptome_assembly/trimmed_fastq/KRWTD1_2_val_2.fq.gz ........... >> merged_R2.fastq
Trinity run
htop
cd ~/rnaseq/transcriptome_assembly
nano trinity.sh
#!/bin/bash
#SBATCH --job-name=<TRINITY>
#SBATCH --cpus-per-task=64
#SBATCH --time=2:00:00
#SBATCH --mem=100g
#SBATCH --mail-type=begin
#SBATCH --mail-type=end
#SBATCH --mail-user=<YOUR ID>@unr.edu
#SBATCH -o <TRINITY>.out # STDOUT
#SBATCH -e <TRINITY>.err # STDERR
Trinity --seqType fq --CPU 64 --max_memory 100G --left merged_R1.fastq --right merged_R2.fastq
Submit job
chmod 775 trinity.sh
MultiQC
cd ~/rnaseq/
multiqc . -n rnaseq_data --exclude rsem --exclude gatk
Please send me the results
mv rnaseq_data.html <your_name>_rnaseq_data.html
Use SCP for downloading and send me the file