Visualization
http://software.broadinstitute.org/software/igv/
The Integrative Genomics Viewer (IGV) is a high-performance visualization tool for interactive exploration of large, integrated genomic datasets. It supports a wide variety of data types, including array-based and next-generation sequence data, and genomic annotations.
IGV is available in multiple forms, including:
the original IGV - a Java desktop application, IGV-Web - a web application, igv.js - a JavaScript component that can be embedded in web pages (for developers)
http://software.broadinstitute.org/software/igv/download
cd /data/gpfs/assoc/bch709/<YOUID>/Genome_assembly/Pilon
mkdir PrePilon
mkdir PostPilon
cp canu_illumina_pilon_sort.bam canu_illumina_pilon_sort.bam.bai canu.illumina.fasta canu.illumina.fasta.fai PostPilon
cp canu_illumina_sort.bam canu_illumina_sort.bam.bai canu.contigs.fasta canu.contigs.fasta.fai PrePilon
Local computer Download For Visualization
canu_illumina_pilon_sort.bam canu_illumina_pilon_sort.bam.bai canu.illumina.fasta canu.illumina.fasta.fai
canu_illumina_sort.bam canu_illumina_sort.bam.bai canu.contigs.fasta canu.contigs.fasta.fai
conda activate postprocess
conda install -c bioconda -c conda-forge htslib=1.9
bgzip -@ 2 canu.illumina.vcf
tabix -p vcf canu.illumina.vcf.gz
Investigate taxa
Here we introduce a software called Kraken2. This tool uses k-mers to assign a taxonomic labels in form of NCBI Taxonomy to the sequence (if possible). The taxonomic label is assigned based on similar k-mer content of the sequence in question to the k-mer content of reference genome sequence. The result is a classification of the sequence in question to the most likely taxonomic label. If the k-mer content is not similar to any genomic sequence in the database used, it will not assign any taxonomic label.
cd /data/gpfs/assoc/bch709/<YOURID>/Genome_assembly/
mkdir taxa
cd taxa
conda deactivate
conda create -n taxa -y python=3.6
conda activate taxa
conda install -c r -c conda-forge -c anaconda -c bioconda kraken kraken2 -y
We can also use another tool by the same group called Centrifuge. This tool uses a novel indexing scheme based on the Burrows-Wheeler transform (BWT) and the Ferragina-Manzini (FM) index, optimized specifically for the metagenomic classification problem to assign a taxonomic labels in form of NCBI Taxonomy to the sequence (if possible). The result is a classification of the sequence in question to the most likely taxonomic label. If the search sequence is not similar to any genomic sequence in the database used, it will not assign any taxonomic label.
conda install -c bioconda centrifuge -y
#!/bin/bash
#SBATCH --job-name=centrifuge
#SBATCH --cpus-per-task=24
#SBATCH --time=12:00:00
#SBATCH --mem=220g
#SBATCH --mail-type=all
#SBATCH --mail-user=wyim@unr.edu
#SBATCH -o centrifuge.out # STDOUT
#SBATCH -e centrifuge.err # STDERR
#SBATCH -p cpu-s2-core-0
#SBATCH -A cpu-s2-bch709-0
centrifuge -x /data/gpfs/assoc/bch709/Course_material/2020/taxa/nt -1 /data/gpfs/assoc/bch709/<YOURID>/Genome_assembly/Illumina/trimmed_fastq/WGS_R1_val_1.fq -2 /data/gpfs/assoc/bch709/<YOURID>/Genome_assembly/Illumina/trimmed_fastq/WGS_R2_val_2.fq --report-file taxa.illumina --threads 24
Centrifuge report
https://fbreitwieser.shinyapps.io/pavian/
#!/bin/bash
#SBATCH --job-name=centrifuge
#SBATCH --cpus-per-task=1
#SBATCH --time=12:00:00
#SBATCH --mem=10g
#SBATCH --mail-type=all
#SBATCH --mail-user=wyim@unr.edu
#SBATCH -o centrifuge.out # STDOUT
#SBATCH -e centrifuge.err # STDERR
#SBATCH -p cpu-s2-core-0
#SBATCH -A cpu-s2-bch709-0
centrifuge-kreport -x /data/gpfs/assoc/bch709/Course_material/2020/taxa/nt taxa.illumina > taxa.illumina.pavian
BUSCO
BUSCO assessments are implemented in open-source software, with a large selection of lineage-specific sets of Benchmarking Universal Single-Copy Orthologs. These conserved orthologs are ideal candidates for large-scale phylogenomics studies, and the annotated BUSCO gene models built during genome assessments provide a comprehensive gene predictor training set for use as part of genome annotation pipelines.
https://busco.ezlab.org/
cd /data/gpfs/assoc/bch709/<YOURID>/Genome_assembly/
mkdir BUSCO
cd BUSCO
cp /data/gpfs/assoc/bch709/<YOURID>/Genome_assembly/genomeassembly_results/*.fasta .
cp /data/gpfs/assoc/bch709/<YOURID>/Genome_assembly/Pilon/canu.illumina.fasta .
conda create -n busco4 python=3.6
conda activate busco4
conda install -c bioconda -c conda-forge busco=4.0.5 multiqc biopython
#!/bin/bash
#SBATCH --job-name=busco
#SBATCH --cpus-per-task=24
#SBATCH --time=12:00:00
#SBATCH --mem=20g
#SBATCH --mail-type=all
#SBATCH --mail-user=wyim@unr.edu
#SBATCH -o busco.out # STDOUT
#SBATCH -e busco.err # STDERR
#SBATCH -p cpu-s2-core-0
#SBATCH -A cpu-s2-bch709-0
#export AUGUSTUS_CONFIG_PATH="~/miniconda3/envs/busco4/config/"
busco -l viridiplantae_odb10 --cpu 24 --in spades_illumina.fasta --out BUSCO_Illumina --mode genome -f
busco -l viridiplantae_odb10 --cpu 24 --in spades_pacbio_illumina.fasta --out BUSCO_Illumina_Pacbio --mode genome -f
busco -l viridiplantae_odb10 --cpu 24 --in canu.contigs.fasta --out BUSCO_Pacbio --mode genome -f
busco -l viridiplantae_odb10 --cpu 24 --in canu.illumina.fasta --out BUSCO_Pacbio_Pilon --mode genome -f
multiqc . -n assembly
BUSCO results
INFO: Results: C:10.8%[S:10.8%,D:0.0%],F:0.5%,M:88.7%,n:425
INFO:
--------------------------------------------------
|Results from dataset viridiplantae_odb10 |
--------------------------------------------------
|C:10.8%[S:10.8%,D:0.0%],F:0.5%,M:88.7%,n:425 |
|46 Complete BUSCOs (C) |
|46 Complete and single-copy BUSCOs (S) |
|0 Complete and duplicated BUSCOs (D) |
|2 Fragmented BUSCOs (F) |
|377 Missing BUSCOs (M) |
|425 Total BUSCO groups searched |
--------------------------------------------------
INFO: BUSCO analysis done. Total running time: 123 seconds
mkdir BUSCO_result
cp BUSCO_*/*.txt BUSCO_result
generate_plot.py -wd BUSCO_result
Chromosome assembly
cd /data/gpfs/assoc/bch709/<YOURID>/Genome_assembly/
mkdir /data/gpfs/assoc/bch709/<YOURID>/Genome_assembly/hic ## will go to genome assembly folder adn make hic folder
cd !$
How can we improve these genome assemblies?
Mate Pair Sequencing
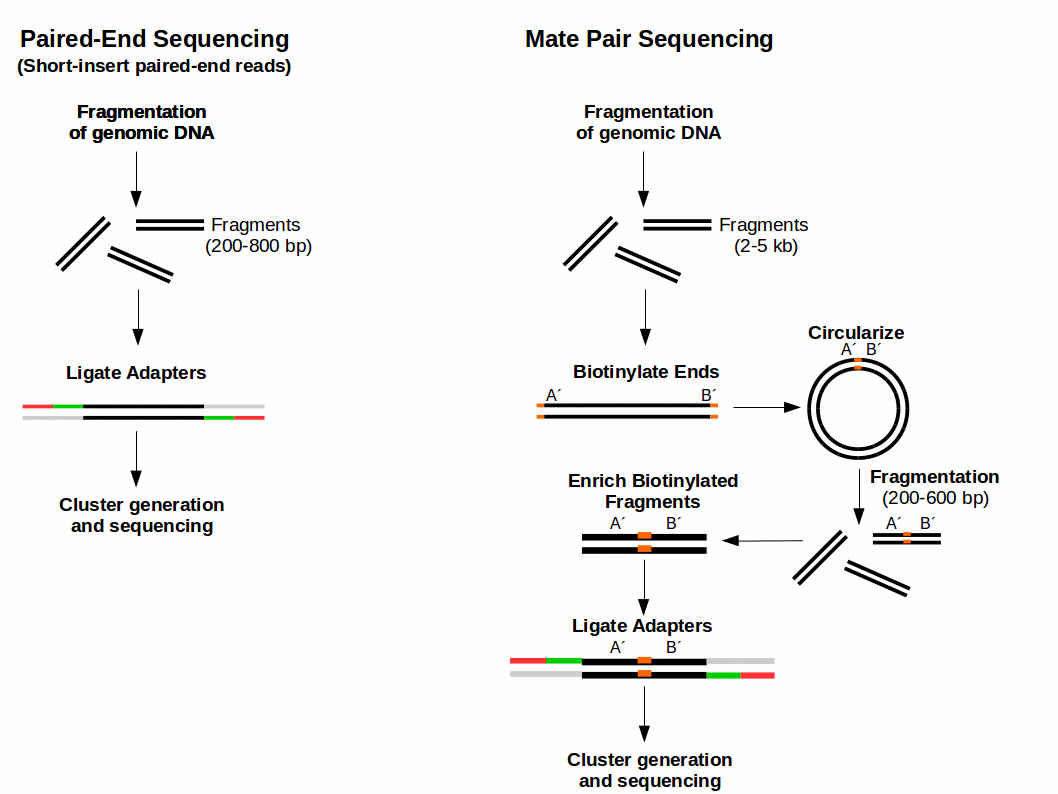
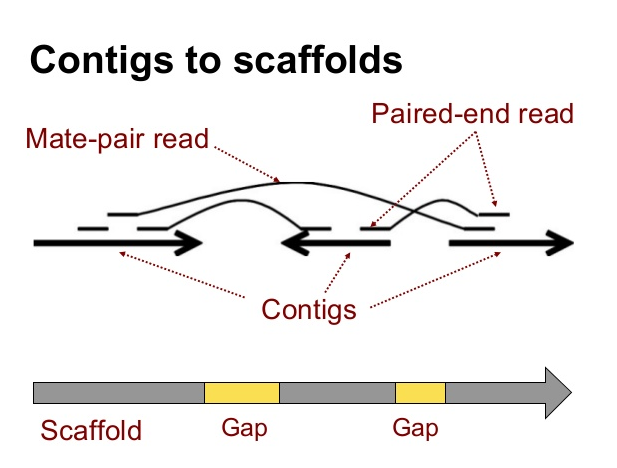
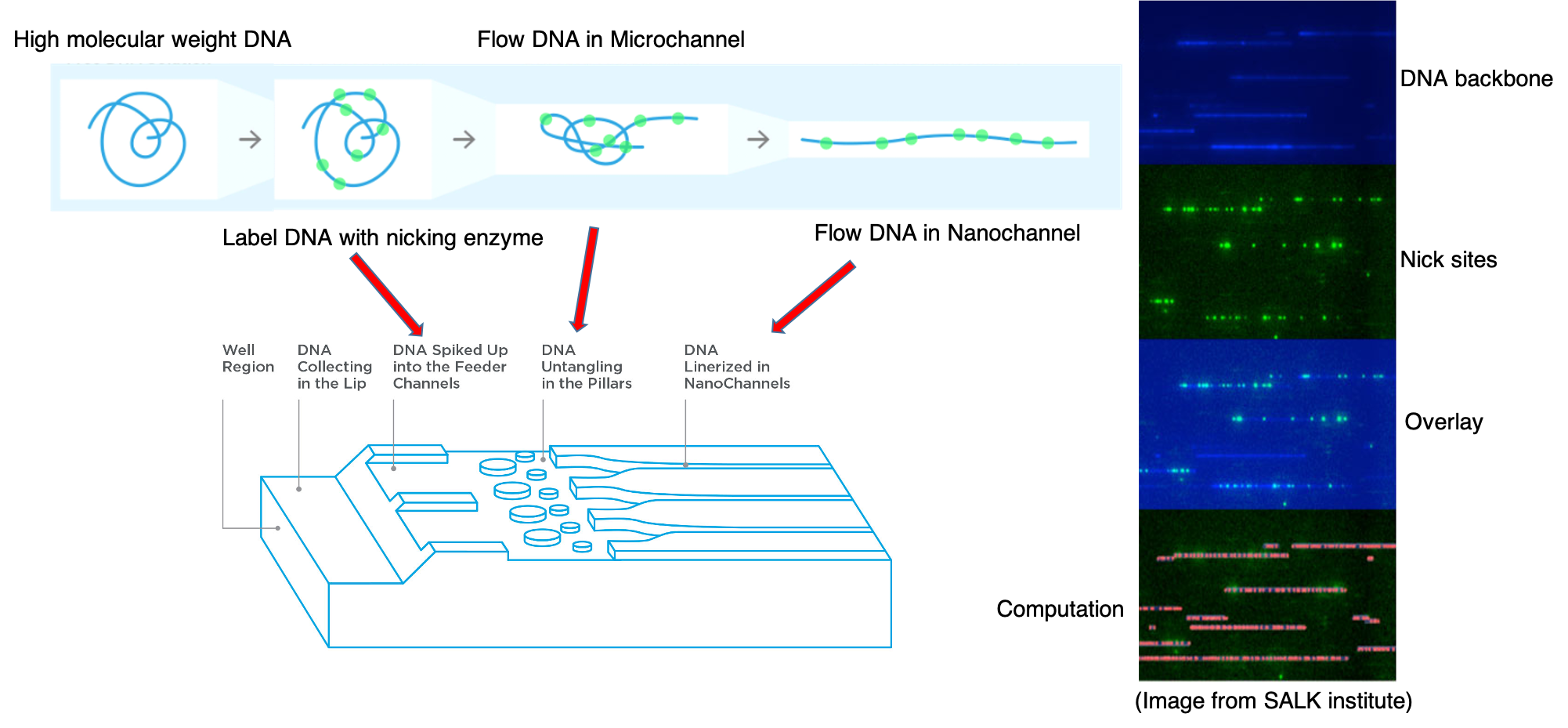
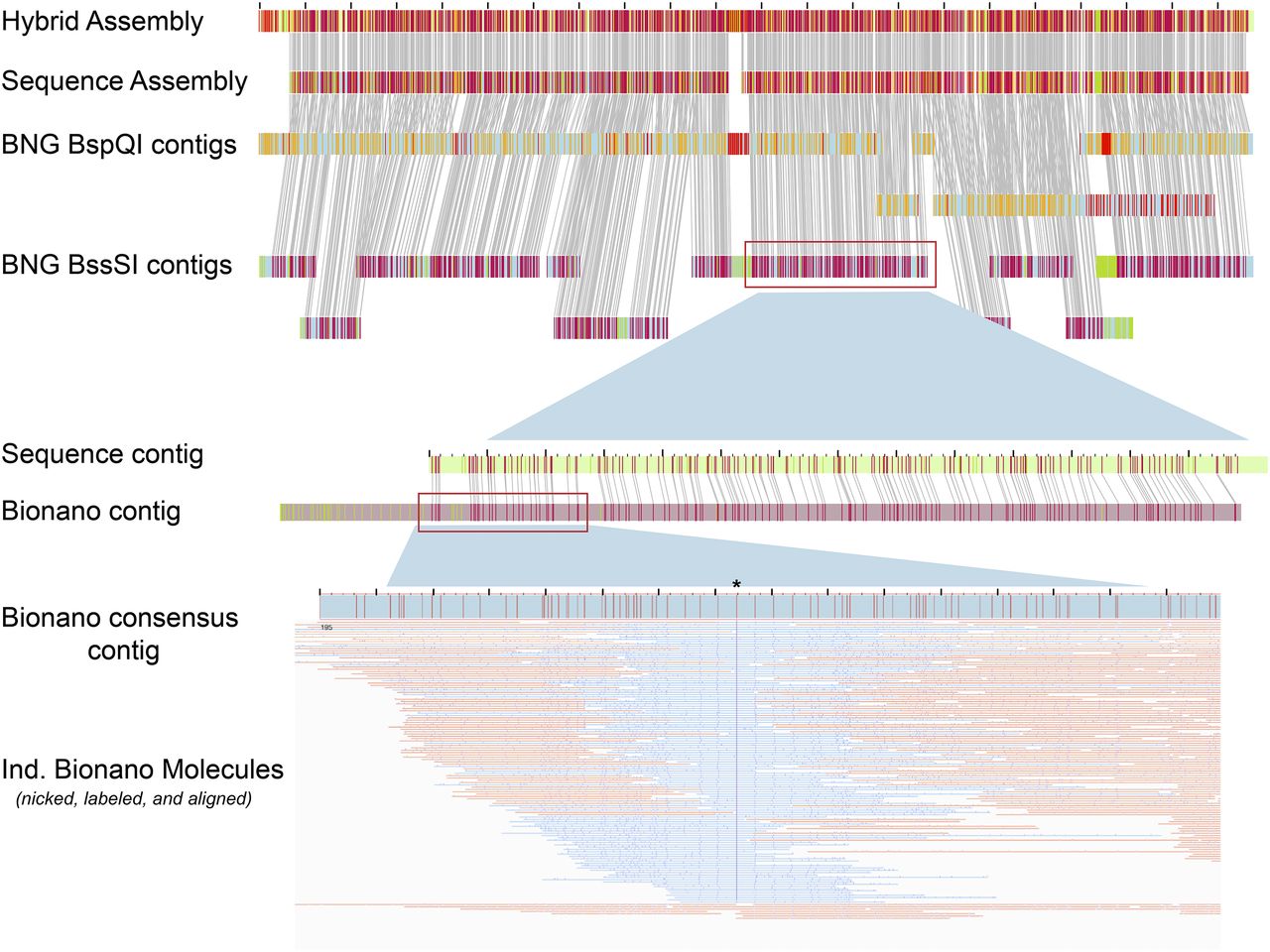
BioNano Optical Mapping
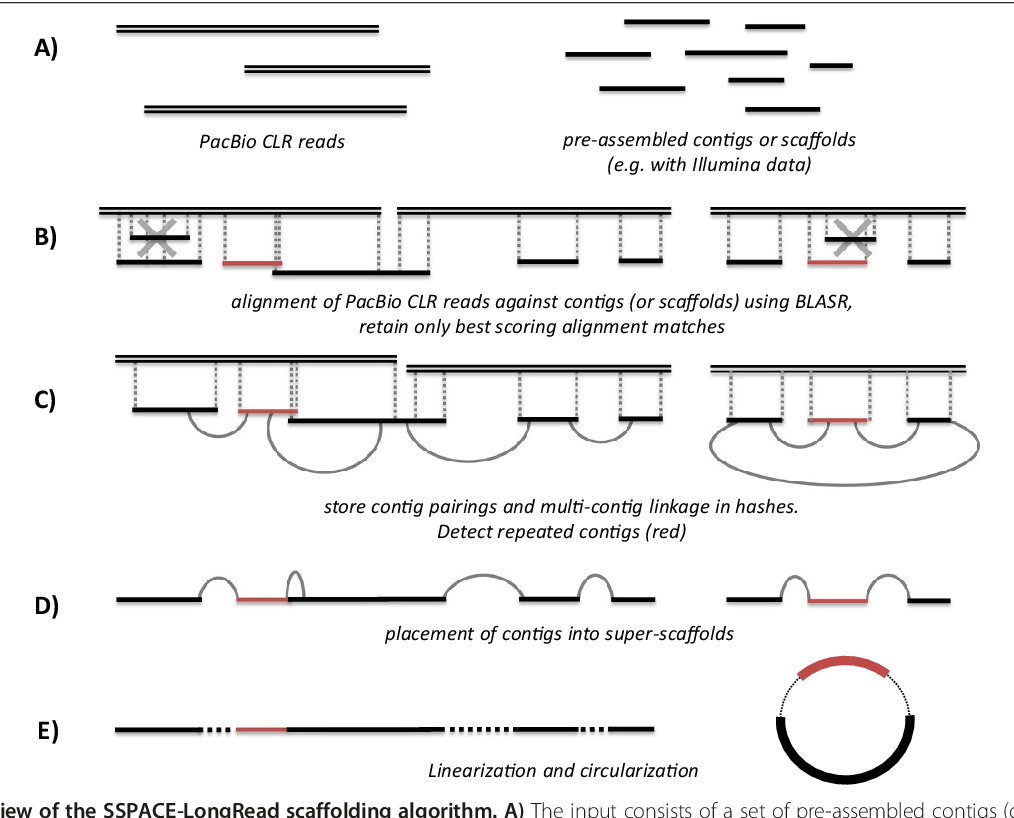
Long Read Scaffolding
Chromosome Conformation Scaffolding