Paper reading
Please read this paper https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-0881-8
RNA Sequencing

- The transcriptome is spatially and temporally dynamic
- Data comes from functional units (coding regions)
- Only a tiny fraction of the genome
Introduction
Sequence based assays of transcriptomes (RNA-seq) are in wide use because of their favorable properties for quantification, transcript discovery and splice isoform identification, as well as adaptability for numerous more specialized measurements. RNA-Seq studies present some challenges that are shared with prior methods such as microarrays and SAGE tagging, and they also present new ones that are specific to high-throughput sequencing platforms and the data they produce. This document is part of an ongoing effort to provide the community with standards and guidelines that will be updated as RNASeq matures and to highlight unmet challenges. The intent is to revise this document periodically to capture new advances and increasingly consolidate standards and best practices.
RNA-Seq experiments are diverse in their aims and design goals, currently including multiple types of RNA isolated from whole cells or from specific sub-cellular compartments or biochemical classes, such as total polyA+ RNA, polysomal RNA, nuclear ribosome-depleted RNA, various size fractions of RNA and a host of others. The goals of individual experiments range from major transcriptome “discovery” that seeks to define and quantify all RNA species in a starting RNA sample to experiments that simply need to detect significant changes in the more abundant RNA classes across many samples.

Seven stages to data science
- Define the question of interest
- Get the data
- Clean the data
- Explore the data
- Fit statistical models
- Communicate the results
- Make your analysis reproducible
What do we need to prepare ?
Sample Information
a. What kind of material it is should be noted: Tissue, cell line, primary cell type, etc… b. It’s ontology term (a DCC wrangler will work with you to obtain this) c. If any treatments or genetic modifications (TALENs, CRISPR, etc…) were done to the sample prior to RNA isolation. d. If it’s a subcellular fraction or derived from another sample. If derived from another sample, that relationship should be noted. e. Some sense of sample abundance: RNA-Seq data from “bulk” vs. 10,000 cell equivalents can give very different results, with lower input samples typically being less reproducible. Having a sense of the amount of starting material here is useful. f. If you received a batch of primary or immortalized cells, the lot #, cat # and supplier should be noted. g. If cells were cultured out, the protocol and methods used to propagate the cells should be noted. h. If any cell phenotyping or other characterizations were done to confirm it’s identify, purity, etc.. those methods should be noted.
RNA Information:
RNAs come in all shapes and sizes. Some of the key properties to report are: a. Total RNA, Poly-A(+) RNA, Poly-A(-) RNA b. Size of the RNA fraction: we typically have a + 200 and – 200 cutoff, but there is a wide range, i.e. microRNA-sized, etc… c. If the RNA was treated with Ribosomal RNA depletion kits (RiboMinus, RiboZero): please note the kit used.
Protocols:
There are several methods used to isolate RNAs with that work fine for the purposes of RNA-Seq. For all the ENCODE libraries that we make, we provide a document that lists in detail: a. The RNA isolation methods, b. Methods of size selections c. Methods of rRNA removal d. Methods of oligo-dT selections e. Methods of DNAse I treatments
Experimental Design
- Balanced design
- Technical replicates not necessary (Marioni et al., 2008)
- Biological replicates: 6 - 12 (Schurch et al., 2016)
- Power analysis
Reading materials
Paul L. Auer and R. W. Doerge “Statistical Design and Analysis of RNA Sequencing Data” Genetics June 1, 2010 vol. 185 no.2 405-416
Busby, Michele A., et al. “Scotty: a web tool for designing RNA-Seq experiments to measure differential gene expression.” Bioinformatics 29.5 (2013): 656-657
Marioni, John C., et al. “RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays.” Genome research (2008)
Schurch, Nicholas J., et al. “How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use?.” Rna (2016)
Zhao, Shilin, et al. “RnaSeqSampleSize: real data based sample size estimation for RNA sequencing.” BMC bioinformatics 19.1 (2018): 191
Replicate number
In all cases, experiments should be performed with two or more biological replicates, unless there is a compelling reason why this is impractical or wasteful (e.g. overlapping time points with high temporal resolution). A biological replicate is defined as an independent growth of cells/tissue and subsequent analysis. Technical replicates from the same RNA library are not required, except to evaluate cases where biological variability is abnormally high. In such instances, separating technical and biological variation is critical. In general, detecting and quantifying low prevalence RNAs is inherently more variable than high abundance RNAs. As part of the ENCODE pipeline, annotated transcript and genes are quantified using RSEM and the values are made available for downstream correlation analysis. Replicate concordance: the gene level quantification should have a Spearman correlation of >0.9 between isogenic replicates and >0.8 between anisogenic replicates.
RNA extraction
- Sample processing and storage
- Total RNA/mRNA/small RNA
- DNAse treatment
- Quantity & quality
- RIN values (Strong effect)
- Batch effect
- Extraction method bias (GC bias)
Reading materials
Romero, Irene Gallego, et al. “RNA-seq: impact of RNA degradation on transcript quantification.” BMC biology 12.1 (2014): 42
Kim, Young-Kook, et al. “Short structured RNAs with low GC content are selectively lost during extraction from a small number of cells.” Molecular cell 46.6 (2012): 893-89500481-9).
RNA Quantification and Quality Control: When working with bulk samples, throughout the various steps we periodically assess the quality and quantity of the RNA. This is typically done on a BioAnalyzer. Points to check are: a. Total RNA b. After oligo-dT size selections c. After rRNA-depletions d. After library construction
Library prep
- PolyA selection
- rRNA depletion
- Size selection
- PCR amplification (See section PCR duplicates)
- Stranded (directional) libraries
- Accurately identify sense/antisense transcript
- Resolve overlapping genes
- Exome capture
- Library normalisation
- Batch effect
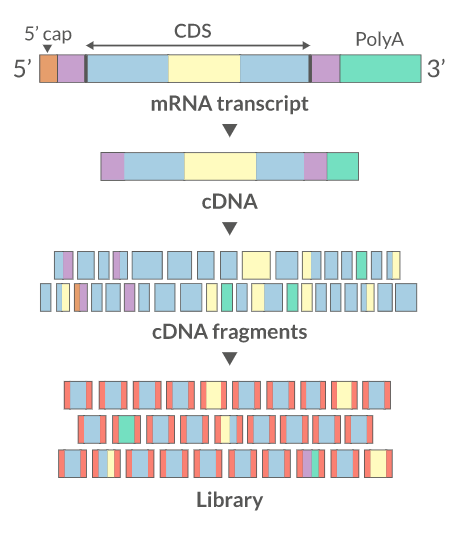

Sequencing:
There are several sequencing platforms and technologies out there being used. It is important to provide the following pieces of information: a. Platform: Illumina, PacBio, Oxford Nanopore, etc… b. Format: Single-end, Pair-end, c. Read Length: 101 bases, 125 bases, etc… d. Unusual barcode placement and sequence: Some protocols introduce barcodes in noncustomary places. If you are going to deliver a FASTQ file that will contain the barcode sequences in it or other molecular markers – you will need to report both the position in the read(s) where they are and their sequence(s). e. Please provide the sequence of any custom primers that were used to sequence the library
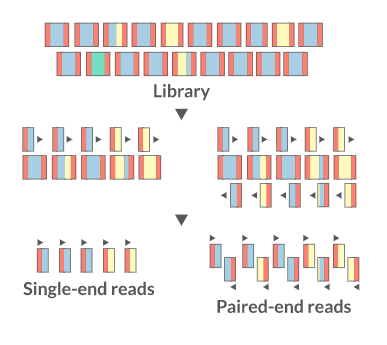
Sequencing depth.
The amount of sequencing needed for a given sample is determined by the goals of the experiment and
the nature of the RNA sample. Experiments whose purpose is to evaluate the similarity between the
transcriptional profiles of two polyA+ samples may require only modest depths of sequencing.
Experiments whose purpose is discovery of novel transcribed elements and strong quantification of
known transcript isoforms requires more extensive sequencing.
• Each Long RNA-Seq library must have a minimum of 30 million aligned reads/mate-pairs.
• Each RAMPAGE library must have a minimum of 20 million aligned reads/mate-pairs.
• Each small RNA-Seq library must have a minimum of 30 million aligned reads/mate-pairs.
Quantitative Standards (spike-ins).
It is highly desirable to include a ladder of RNA spike-ins to calibrate quantification, sensitivity, coverage and linearity. Information about the spikes should include the stage of sample preparation that the spiked controls were added, as the point of entry affects use of spike data in the output. In general, introducing spike-ins as early in the process as possible is the goal, with more elaborate uses of different spikes at different steps being optional (e.g. before poly A+ selection, at the time of cDNA synthesis, or just prior to sequencing). Different spike-in controls are needed for each of the RNA types being analyzed (e.g. long RNAs require different quantitative controls from short RNAs). Such standards are not yet available for all RNA types. Information about quantified standards should also include: a) A FASTA (or other standard format) file containing the sequences of each spike in. b) Source of the spike-ins (home-made, Ambion, etc..) c) The concentration of each of the spike-ins in the pool used.
Hong et al., 2016, Principles of metadata organization at the ENCODE data coordination center.
QC FAIL?
https://sequencing.qcfail.com/
Fastq format
FASTQ format is a text-based format for storing both a biological sequence (usually nucleotide sequence) and its corresponding quality scores.
The format is similar to fasta though there are differences in syntax as well as integration of quality scores. Each sequence requires at least 4 lines:
- The first line is the sequence header which starts with an ‘@’ (not a ‘>’!). Everything from the leading ‘@’ to the first whitespace character is considered the sequence identifier. Everything after the first space is considered the sequence description
- The second line is the sequence.
- The third line starts with ‘+’ and can have the same sequence identifier appended (but usually doesn’t anymore).
- The fourth line are the quality scores
The FastQ sequence identifier generally adheres to a particular format, all of which is information related to the sequencer and its position on the flowcell. The sequence description also follows a particular format and holds information regarding sample information.
$ pwd
$ cd ~/
$ mkdir bch709/rnaseq
$ cd bch709/rnaseq/
$ pwd
$ wget https://www.dropbox.com/s/y7yehmfze1l6cgz/pair1.fastq.gz
$ wget https://www.dropbox.com/s/xsrth6icapyr4p0/pair2.fastq.gz
$ ls -algh
$ zcat pair2.fastq.gz | head
@A00261:180:HL7GCDSXX:2:1101:30572:1047/2
AAAATACATTGATGACCATCTAAAGTCTACGGCGTATGCGACTGATGAAGTATATTGCACCACCTGAGGGTGATGCTAATACTACTGTTGACGATAATGCTGATCTTCTTGCTAAGCTTAATATTGTTGGTGTTGAACCTAATGTTGGTG
+
FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFFFFFFFF
@A00261:180:HL7GCDSXX:2:1101:21088:1094/2
ATCTCACATCGTTCCCTCAAGATTCTGAATTTTGGCAGCTCATTGCATTCTGTGCCGGCACTGGTGGTTCGATGCTTGTCATTGGTTCTGCTGCTGGTGTAGCCTTCATGGGGATGGAGAAAGTCGATTTCTTTTGGTATTTCCGAAAGG
+
FFFFFFFFFFFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF
@A00261:180:HL7GCDSXX:2:1101:21251:1125/2
CGGTGGAAAAGGAAACAGCTTTGGAAGGTTGATTCCATTACAGATTCGATTCGAAACTATGGTTCAGATTTCCGATCTTCCACGGGATTTGACAGAGGAGGTGCTCTCTAGGATTCCGGTGACATCTATGAGAGCAGTGAGATTTACTTG
- A00261 : Instrument name
- 180 : run ID
- HL7GCDSXX : Flowcell ID
- 2 : Flowcell lane
- 1101 : tile number within the flowcell lane
- 30572 : X-coordinate of the cluster within tile
- 1047 : Y-coordinate of the cluster within tile
- /2 : member of a pair 1 or 2 (Paired end reads only)
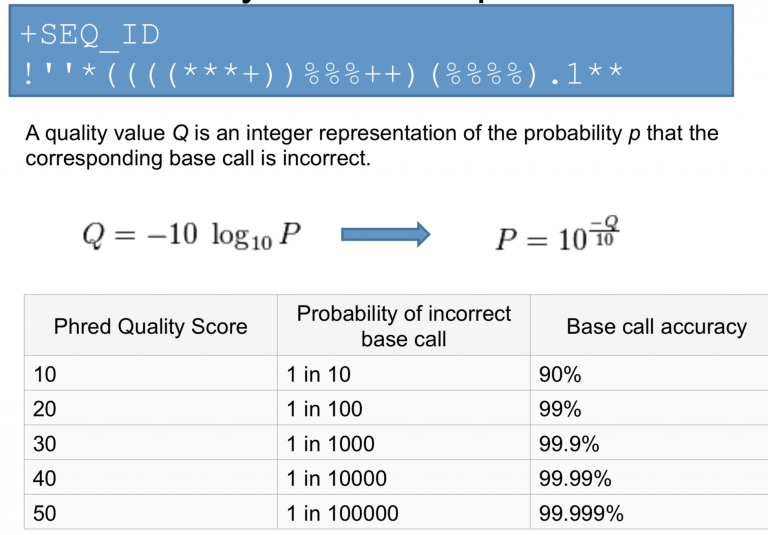
Quality Scores
Quality scores are a way to assign confidence to a particular base within a read. Some sequencers have their own proprietary quality encoding but most have adopted Phred-33 encoding. Each quality score represents the probability of an incorrect basecall at that position.
Phred Quality Score Encoding
Quality scores started as numbers (0-40) but have since changed to an ASCII encoding to reduce filesize and make working with this format a bit easier, however they still hold the same information. ASCII codes are assigned based on the formula found below. This table can serve as a lookup as you progress through your analysis.
Quality Score Interpretation
Once you know what each quality score represents you can then use this chart to understand the confidence in a particular base.
BW algorithm
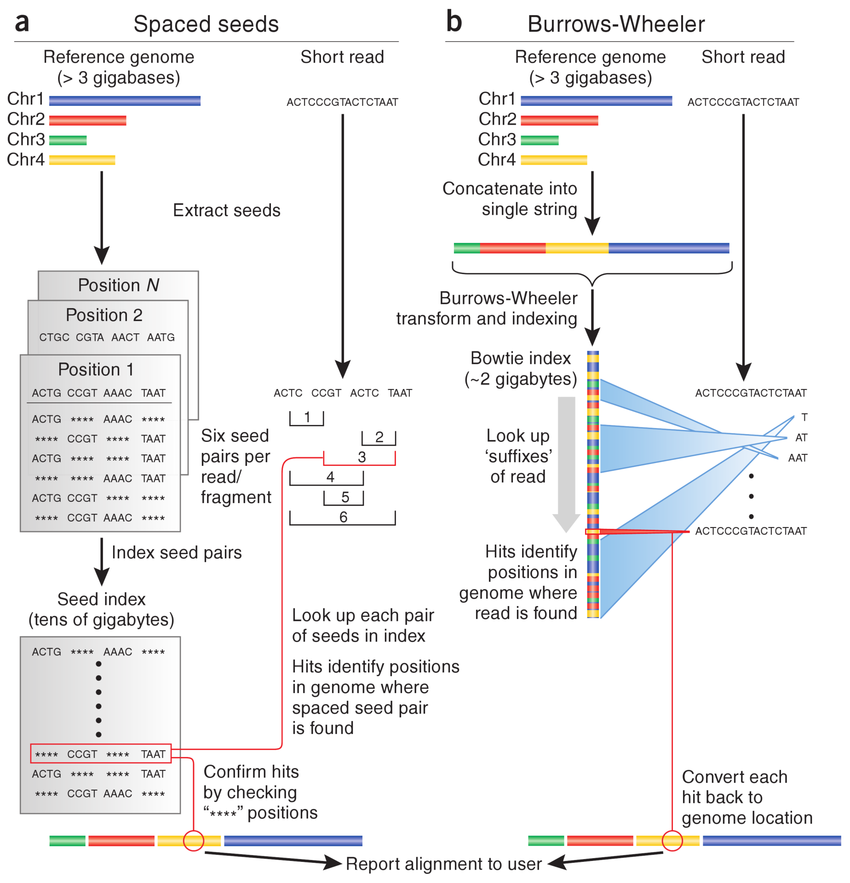

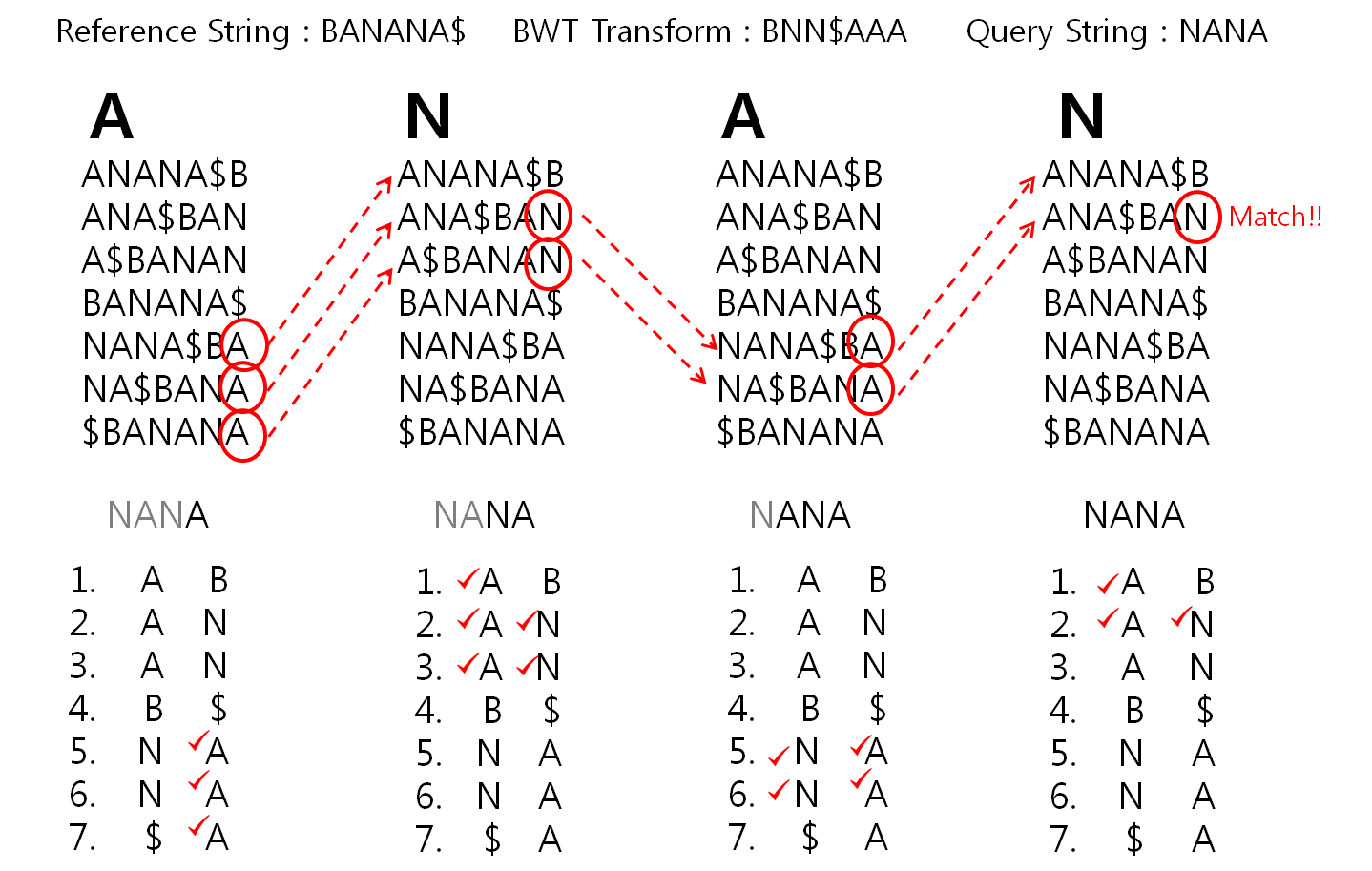
Conda enviroment
conda create -n rnaseq -c bioconda -c conda-forge fastqc trim-galore hisat2 samtools subread bioconductor-deseq2 'multiqc<1.34' -y
Reads QC
- Number of reads
- Per base sequence quality
- Per sequence quality score
- Per base sequence content
- Per sequence GC content
- Per base N content
- Sequence length distribution
- Sequence duplication levels
- Overrepresented sequences
- Adapter content
- Kmer content
Run fastqc
fastqc --help
fastqc -t <YOUR CPU COUNT> paired1.fastq.gz paired2.fastq.gz
How to make a report?

RNASeq tutorial
MacOS JUST ONE TIME Mac terminal
echo 'setopt nonomatch' >> ~/.zshrc
restart terminal
Environment create and installation in Pronghorn
# Create a new conda environment named "rnaseq".
# Add two channels to fetch the required packages:
# - bioconda: A channel specializing in bioinformatics software
# - conda-forge: A community-maintained collection of conda packages
-c bioconda -c conda-forge
# List of packages/software to be installed in the "rnaseq" environment:
# - fastqc: A tool for quality control checks on raw sequence data
# - trim-galore: A wrapper tool around Cutadapt and FastQC to consistently apply adapter and quality trimming
# - hisat2: A fast and sensitive alignment program for mapping next-generation sequencing reads to a population of genomes
# - samtools: A suite of programs for interacting with high-throughput sequencing data
# - subread: A toolkit for processing next-gen sequencing read data, including feature counting
# - bioconductor-deseq2: A package for differential expression analysis based on the negative binomial distribution
# - bc: An arbitrary precision calculator language
# The "-y" flag allows the command to proceed without asking for user confirmation.
conda create -n rnaseq -c bioconda -c conda-forge fastqc trim-galore hisat2 samtools subread bioconductor-deseq2 bc 'multiqc<1.34' -y
Isaac, Sahar, Pavani
mkdir /data/gpfs/assoc/bch709-4/${USER}
mv ~/bch709 ~/bch709_seqkit
ln -s /data/gpfs/assoc/bch709-4/${USER} ~/bch709
cd ~/bch709
# if this is not working follow this
mkdir -p ~/bch709/rnaseq
Move to working path
# Create a directory and its parent directories as needed.
# The "-p" flag ensures that no error will be thrown if the directory already exists,
# and it also allows for the creation of parent directories if they don't exist.
mkdir -p ~/bch709/rnaseq
# Change the current working directory to the newly created directory.
cd ~/bch709/rnaseq
Trim the reads
- Trim IF necessary
- Synthetic bases can be an issue for SNP calling
- Insert size distribution may be more important for assemblers
- Trim/Clip/Filter reads
- Remove adapter sequences
- Trim reads by quality
- Sliding window trimming
- Filter by min/max read length
- Remove reads less than ~18nt
- Demultiplexing/Splitting
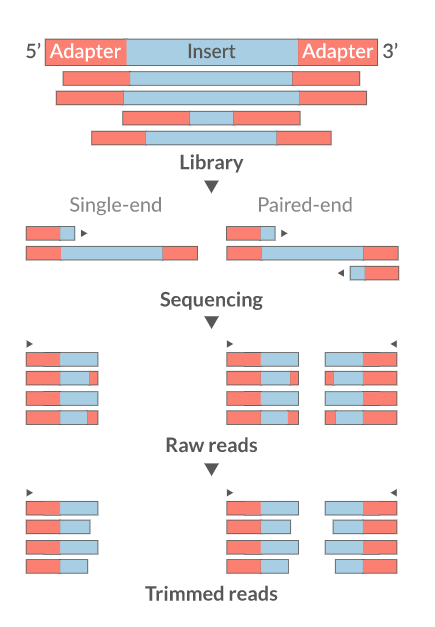
Cutadapt
fastp
Skewer
Prinseq
Trimmomatics
Trim Galore
Activate environment
# Activate a conda environment named "rnaseq".
# By activating an environment, the user can utilize the specific packages and tools
# installed in that environment without affecting other projects or the system-wide installation.
conda activate rnaseq
Run trimming
# Copy the "paired1.fastq.gz" file from the source directory to the "~/bch709/rnaseq" directory.
cp /data/gpfs/assoc/bch709-4/data/paired1.fastq.gz ~/bch709/rnaseq
# Copy the "paired2.fastq.gz" file from the source directory to the "~/bch709/rnaseq" directory.
cp /data/gpfs/assoc/bch709-4/data/paired2.fastq.gz ~/bch709/rnaseq
# Copy the "bch709.fasta" file (contains genomic sequences) from the source directory to the "~/bch709/rnaseq" directory.
cp /data/gpfs/assoc/bch709-4/data/bch709.fasta ~/bch709/rnaseq
# Copy the "bch709.gtf" file (contains genomic annotation) from the source directory to the "~/bch709/rnaseq" directory.
cp /data/gpfs/assoc/bch709-4/data/bch709.gtf ~/bch709/rnaseq
# Change the current working directory to "~/bch709/rnaseq".
cd ~/bch709/rnaseq
# Display the help documentation for "trim_galore", a wrapper tool around Cutadapt and FastQC to consistently apply adapter and quality trimming.
trim_galore --help
# Execute "trim_galore" with specified options:
# --paired: Indicate that the input consists of paired-end reads.
# --three_prime_clip_R1 5: Remove 5 bases from the 3' end of the first read.
# --three_prime_clip_R2 5: Remove 5 bases from the 3' end of the second read.
# --cores 2: Use 2 CPU cores.
# --max_n 40: Remove sequences that contain more than 40 'N' bases.
# --fastqc: Run FastQC in the end to provide quality control checks.
# --gzip: Output files will be compressed using gzip.
# -o trim: Output trimmed files to a directory named "trim".
# The last two arguments specify the input files to be trimmed.
trim_galore --paired --three_prime_clip_R1 5 --three_prime_clip_R2 5 --cores 2 --max_n 40 --fastqc --gzip -o trim paired1.fastq.gz paired2.fastq.gz
# Run "multiqc" to aggregate the results of bioinformatics analyses (like FastQC) into a single report:
# --dirs: Specify the directory where the analysis data can be found.
# --filename: Name of the output report file.
multiqc --dirs ~/bch709/rnaseq --filename trim --exclude rsem --exclude gatk
Download file in your local terminal
Windows
# The 'scp' command (secure copy) is used for securely transferring files between a local and a remote machine over SSH (secure shell).
# Source of the files:
# Replace "YOURID" with your actual username on the remote server "pronghorn.rc.unr.edu".
# Files with the ".html" extension from the directory "~/bch709/rnaseq/" on the remote server will be copied.
scp YOURID@pronghorn.rc.unr.edu:~/bch709/rnaseq/*.html ~/bch709
# This command attempts to open the "bch709" directory located in the home directory
# using the Windows File Explorer.
explorer.exe ~/bch709
MacOS
scp <<<YOURID>>>@pronghorn.rc.unr.edu:~/bch709/rnaseq/*.html ~/bch709
open ~/bch709
Align the reads (mapping)
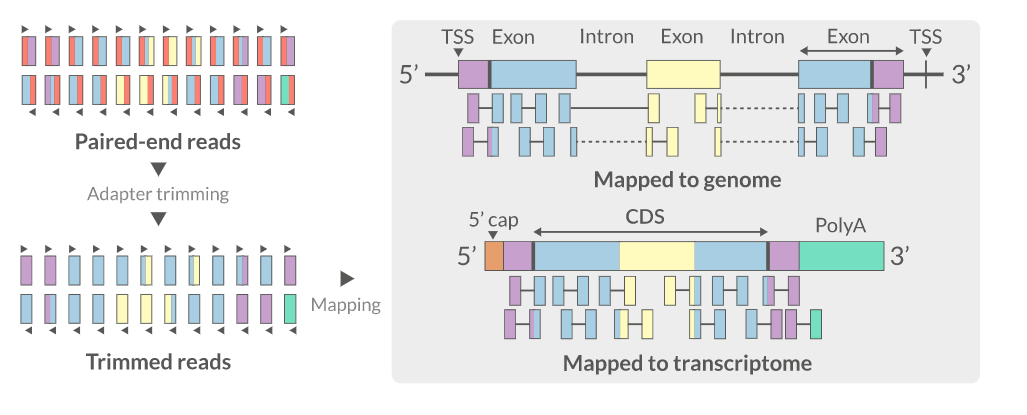
- Aligning reads back to a reference sequence
- Mapping to genome vs transcriptome
- Splice-aware alignment (genome)
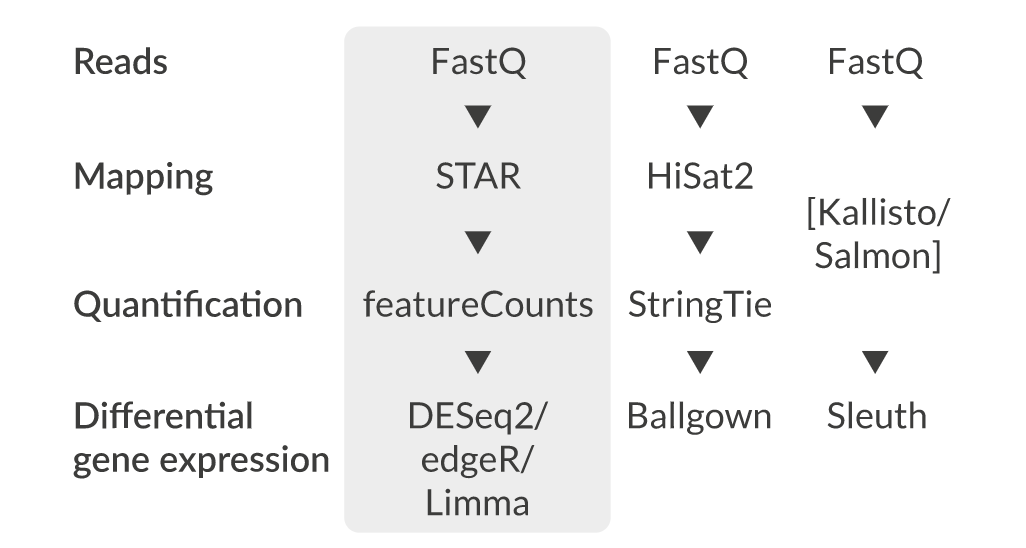
STAR
HISAT2
GSNAP
Bowtie2
Novoalign
HISAT2 (graph FM index, spin off version Burrows-Wheeler Transform)
HISAT2 indexing
# Display the help documentation for the "hisat2-build" command.
# This is useful for understanding the available options and their descriptions.
hisat2-build --help
# Build the index for "hisat2" using the "hisat2-build" command:
# "bch709.fasta" is the input reference genome in FASTA format.
# "bch709" is the prefix for the output index files.
# After execution, several index files will be generated with this prefix, like "bch709.1.ht2", "bch709.2.ht2", etc.
hisat2-build bch709.fasta bch709
HISAT2 mapping
# Execute the 'hisat2' command to perform alignment:
# -x bch709: Specify the prefix of the index files. This tells 'hisat2' to use the previously built index files with the prefix "bch709".
# --threads 2: Use 2 CPU threads for the alignment process, which can speed up the alignment.
# -1 trim/paired1_val_1.fq.gz: Specify the path to the first file of paired-end reads (usually referred to as the "forward" reads).
# -2 trim/paired2_val_2.fq.gz: Specify the path to the second file of paired-end reads (usually referred to as the "reverse" reads).
# -S align.sam: Output the alignment results in SAM format and save it to a file named "align.sam".
# --summary-file alignment.txt: Write a summary of the alignment process to a file named "alignment.txt".
hisat2 -x bch709 --threads 2 -1 trim/paired1_val_1.fq.gz -2 trim/paired2_val_2.fq.gz -S align.sam --summary-file alignment.txt
# Display the contents of the "alignment.txt" file using the 'cat' command.
# This file contains a summary of the alignment process, such as the number of reads aligned successfully,
# number of reads that failed to align, etc.
cat alignment.txt
##
# Run "multiqc" to aggregate the results of bioinformatics analyses (like FastQC) into a single report:
# --dirs: Specify the directory where the analysis data can be found.
# --filename: Name of the output report file.
multiqc --dirs ~/bch709/rnaseq --filename mapping --exclude rsem --exclude gatk
Download file in your local terminal
scp <<<YOURID>>>@pronghorn.rc.unr.edu:~/bch709/rnaseq/*.html .
SAM file format
Check result
head align.sam
SAM file
<QNAME> <FLAG> <RNAME> <POS> <MAPQ> <CIGAR> <MRNM> <MPOS> <ISIZE> <SEQ> <QUAL> [<TAG>:<VTYPE>:<VALUE> [...]]
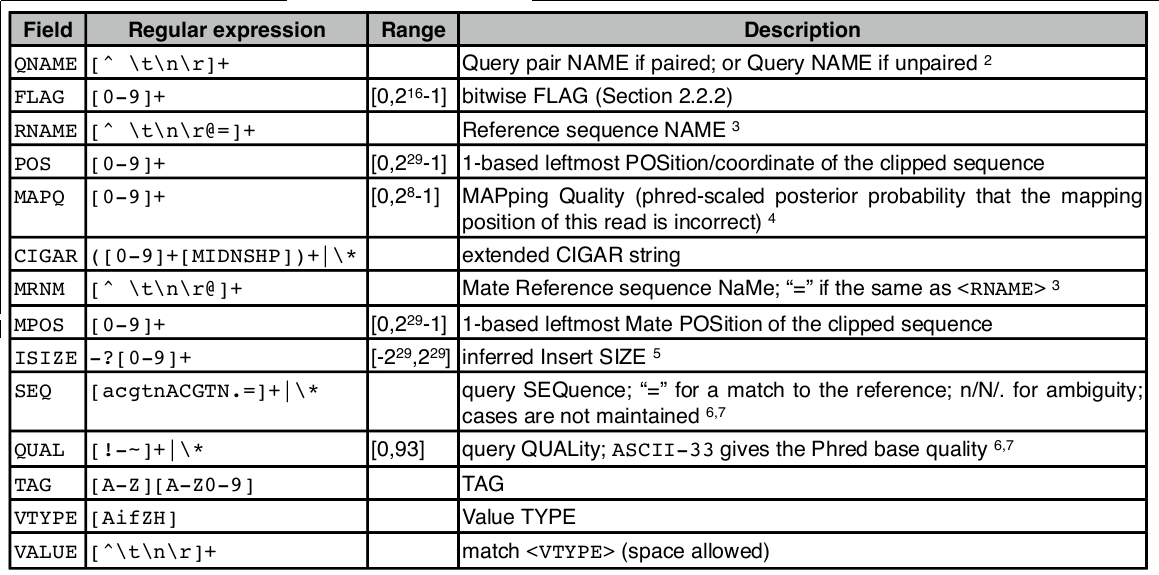
SAM flag

Demical to binary
echo 'obase=163;10' | bc
SAM tag
There are a bunch of predefined tags, please see the SAM manual for more information. For the tags used in this example:
Any tags that start with X? are reserved fields for end users: XT:A:M, XN:i:2, XM:i:0, XO:i:0, XG:i:0
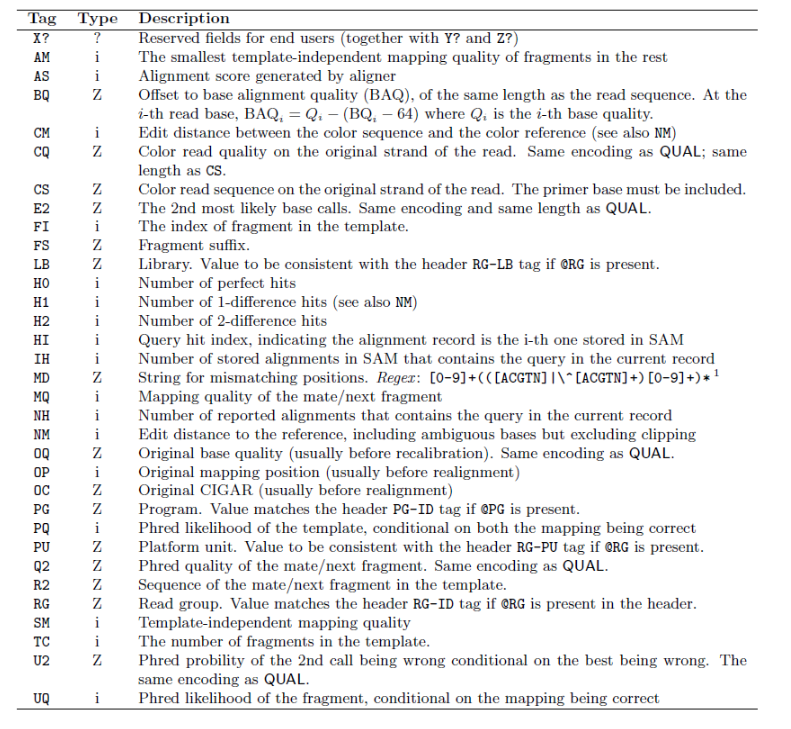
More information is below.
http://samtools.github.io/hts-specs/
SAMtools
SAM (Sequence Alignment/Map) format is a generic format for storing large nucleotide sequence alignments. SAM aims to be a format that:
- Is flexible enough to store all the alignment information generated by various alignment programs;
- Is simple enough to be easily generated by alignment programs or converted from existing alignment formats;
- Is compact in file size;
- Allows most of operations on the alignment to work on a stream without loading the whole alignment into memory;
- Allows the file to be indexed by genomic position to efficiently retrieve all reads aligning to a locus.
SAM Tools provide various utilities for manipulating alignments in the SAM format, including sorting, merging, indexing and generating alignments in a per-position format. http://samtools.sourceforge.net/
# Convert the SAM (Sequence Alignment/Map) format file "align.sam" to BAM (Binary Alignment/Map) format.
# The '-S' flag indicates that the input is in SAM format.
# The '-b' flag specifies that the output should be in BAM format.
# The result is redirected to a file named "align.bam".
samtools view -Sb align.sam > align.bam
# Sort the BAM file "align.bam" by chromosomal position.
# The result will be saved as "align_sort.bam" specified by the '-o' flag.
samtools sort align.bam -o align_sort.bam
# Create an index for the sorted BAM file "align_sort.bam".
# This index file (with a ".bai" extension) allows for quicker access to the BAM file for various operations like viewing, analysis, etc.
samtools index align_sort.bam
# Generate statistics on the sorted BAM file and save the result to "align_sort.bam.stat".
samtools stats align_sort.bam > align_sort.bam.stat
# Display the contents of the "align_sort.bam.stat" file.
# This file contains detailed statistics on the alignment, like total reads, mapped reads, average read length, etc.
cat align_sort.bam.stat
# List the files and directories in the current directory in a long format with human-readable file sizes.
# '-a' shows all entries, including hidden ones.
# '-l' provides a long listing format.
# '-g' omits the owner name.
# '-h' provides human-readable sizes (e.g., 1K, 234M, 2G).
ls -algh
# List the files and directories in the current directory with the same flags as above.
# Additionally, '-t' sorts by modification time, and '-r' reverses the order.
# This results in showing the oldest modified files/directories first.
ls -alghtr
BAM file
A BAM file (.bam) is the binary version of a SAM file. A SAM file (.sam) is a tab-delimited text file that contains sequence alignment data.
| SAM/BAM | size |
|---|---|
| align.sam | 968M |
| align.bam | 86M |
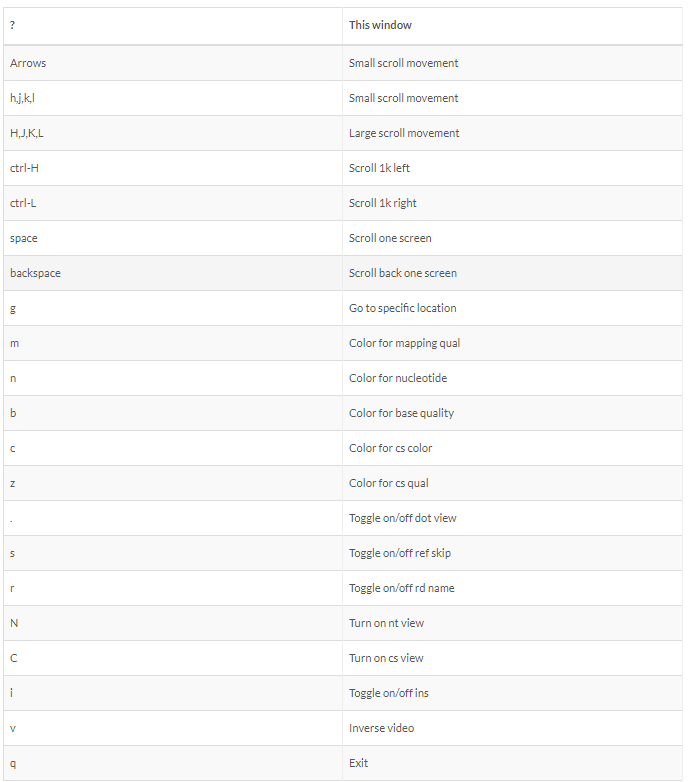
Alignment visualization
COLUMNS=150 samtools tview -d t align_sort.bam bch709.fasta
Alignment QC
multiqc --dirs ~/bch709/rnaseq --filename samtools --exclude rsem --exclude gatk
Download file in your local terminal
MacOS
scp <<<YOURID>>>@pronghorn.rc.unr.edu:~/bch709/rnaseq/*.html ~/bch709
Windows
scp <<<YOURID>>>@pronghorn.rc.unr.edu:~/bch709/rnaseq/*.html ~/bch709
Alignment QC
- Number of reads mapped/unmapped/paired etc
- Uniquely mapped
- Insert size distribution
- Coverage
- Gene body coverage
- Biotype counts / Chromosome counts
- Counts by region: gene/intron/non-genic
- Sequencing saturation
- Strand specificity
samtools > stats
bamtools > stats
QoRTs
RSeQC
Qualimap
Quantification • Counts
- Read counts = gene expression
- Reads can be quantified on any feature (gene, transcript, exon etc)
- Intersection on gene models
- Gene/Transcript level

featureCounts HTSeq RSEM Cufflinks Rcount
Quantification method
- PCR duplicates
- Ignore for RNA-Seq data
- Computational deduplication (Don’t!)
- Use PCR-free library-prep kits
-
Use UMIs during library-prep
- Multi-mapping
- Added (BEDTools multicov)
- Discard (featureCounts, HTSeq)
- Distribute counts (Cufflinks)
- Rescue
- Probabilistic assignment (Rcount, Cufflinks)
- Prioritise features (Rcount)
- Probabilistic assignment with EM (RSEM)
mapping count
conda install -c conda-forge -c bioconda subread
featureCounts -p -a bch709.gtf align_sort.bam -o counts.txt
Reference
Fu, Yu, et al. “Elimination of PCR duplicates in RNA-seq and small RNA-seq using unique molecular identifiers.” BMC genomics 19.1 (2018): 531 Parekh, Swati, et al. “The impact of amplification on differential expression analyses by RNA-seq.” Scientific reports 6 (2016): 25533 Klepikova, Anna V., et al. “Effect of method of deduplication on estimation of differential gene expression using RNA-seq.” PeerJ 5 (2017): e3091
Differential expression
DESeq2 edgeR (Neg-binom > GLM > Test) Limma-Voom (Neg-binom > Voom-transform > LM > Test)
Functional analysis • GO
Gene enrichment analysis (Hypergeometric test) Gene set enrichment analysis (GSEA) Gene ontology / Reactome databases
Reading material
Conesa, Ana, et al. “A survey of best practices for RNA-seq data analysis.” Genome biology 17.1 (2016): 13
Reference:
- Conda documentation https://docs.conda.io/en/latest/
- Conda-forge https://conda-forge.github.io/
- BioConda https://bioconda.github.io/