Location
mkdir -p ~/bch709/BLAST
cd ~/bch709/BLAST
ENV
conda create -n blast -c bioconda -c conda-forge blast seqkit -y
BLAST
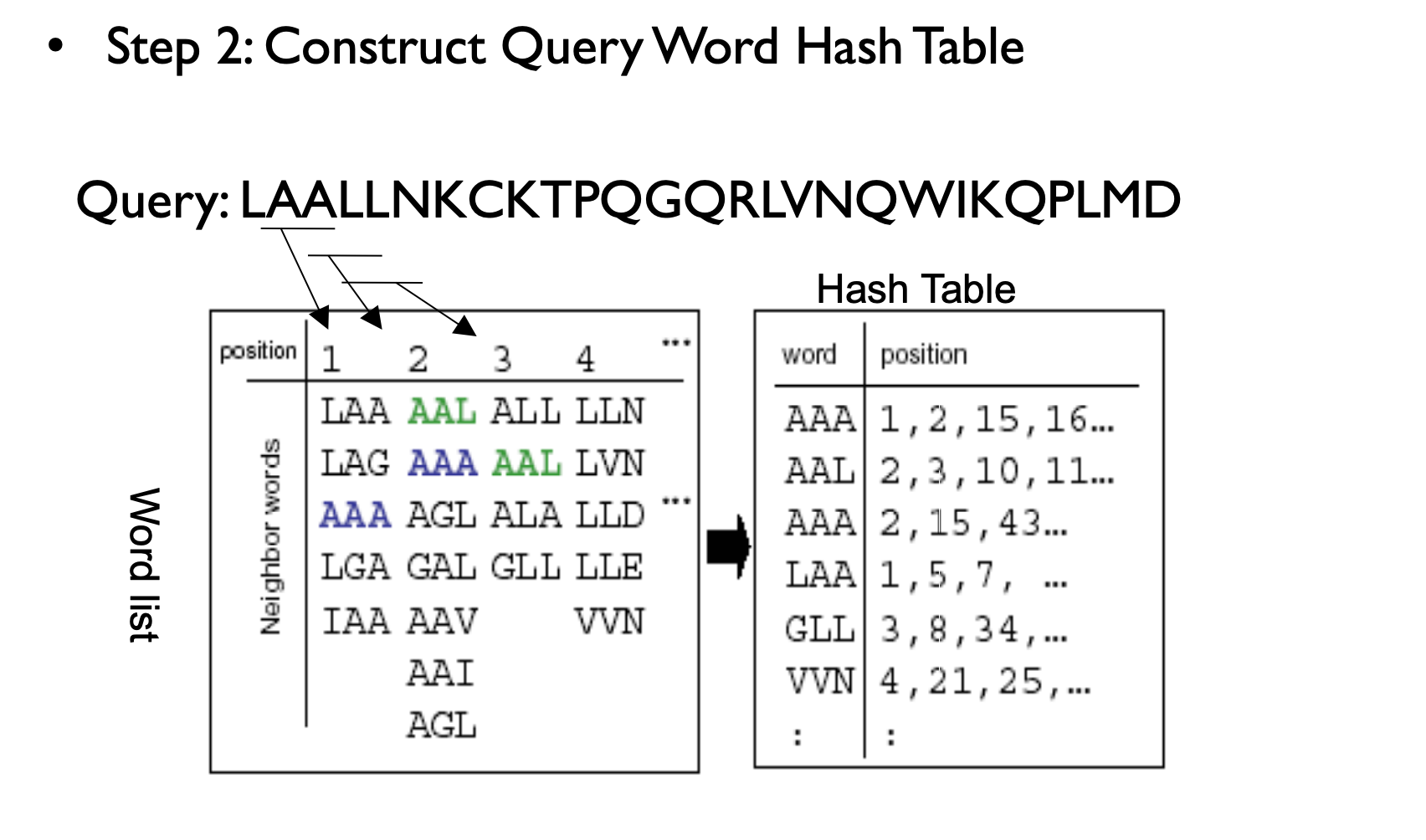
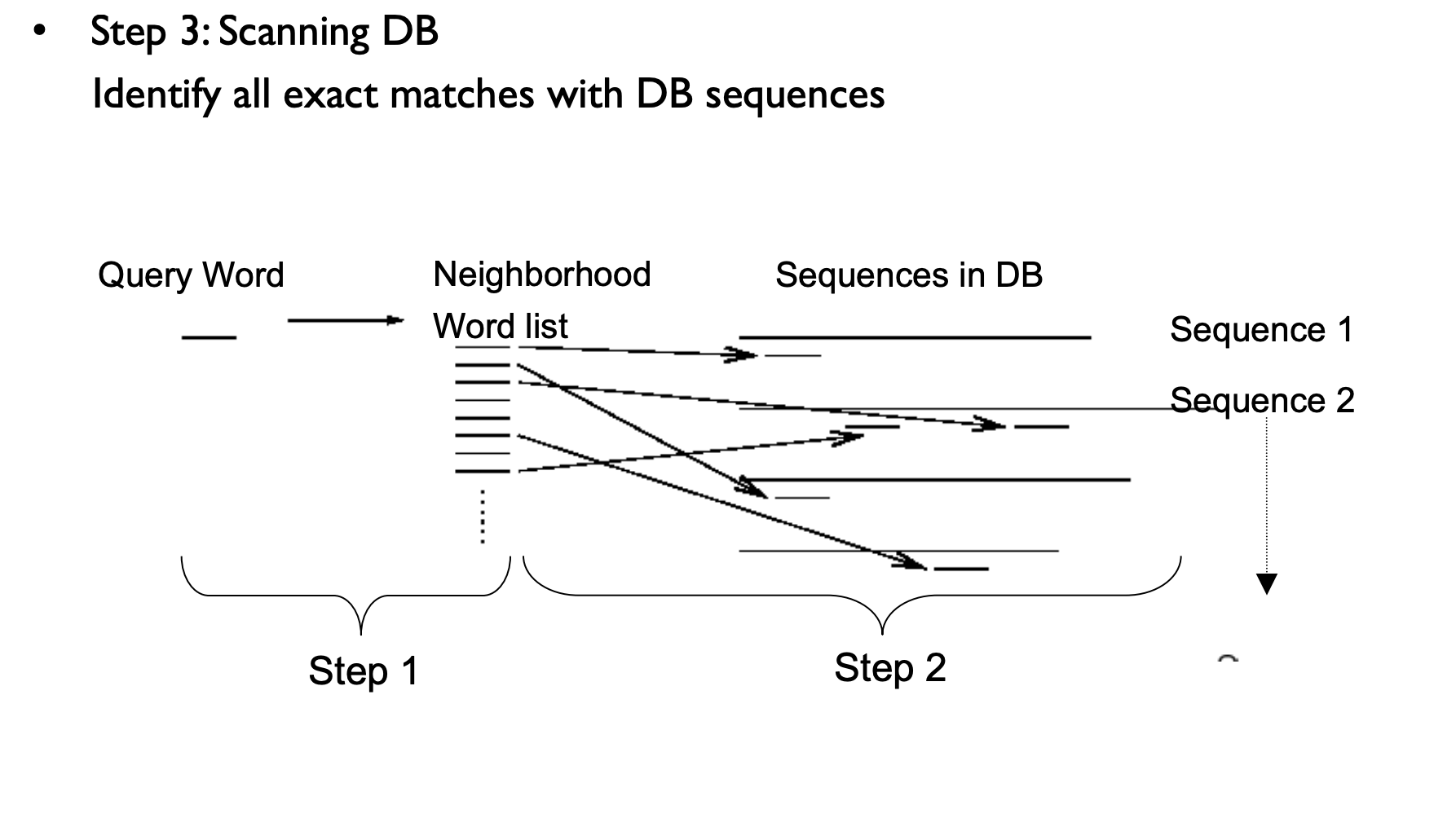
Basic Local Alignment Search Tool (Altschul et al., 1990 & 1997) is a sequence comparison algorithm optimized for speed used to search sequence databases for optimal local alignments to a query. The initial search is done for a word of length “W” that scores at least “T” when compared to the query using a substitution matrix. Word hits are then extended in either direction in an attempt to generate an alignment with a score exceeding the threshold of “S”. The “T” parameter dictates the speed and sensitivity of the search.
Rapidly compare a sequence Q to a database to find all sequences in the database with an score above some cutoff S.
- Which protein is most similar to a newly sequenced one?
- Where does this sequence of DNA originate?
- Speed achieved by using a procedure that typically finds most matches with scores > S.
- Tradeoff between sensitivity and specificity/speed
- Sensitivity – ability to find all related sequences
- Specificity – ability to reject unrelated sequences
Homologous sequence are likely to contain a short high scoring word pair, a seed.
– Unlike Baeza-Yates, BLAST doesn’t make explicit guarantees
BLAST then tries to extend high scoring word pairs to compute maximal high scoring segment pairs (HSPs).
– Heuristic algorithm but evaluates the result statistically.
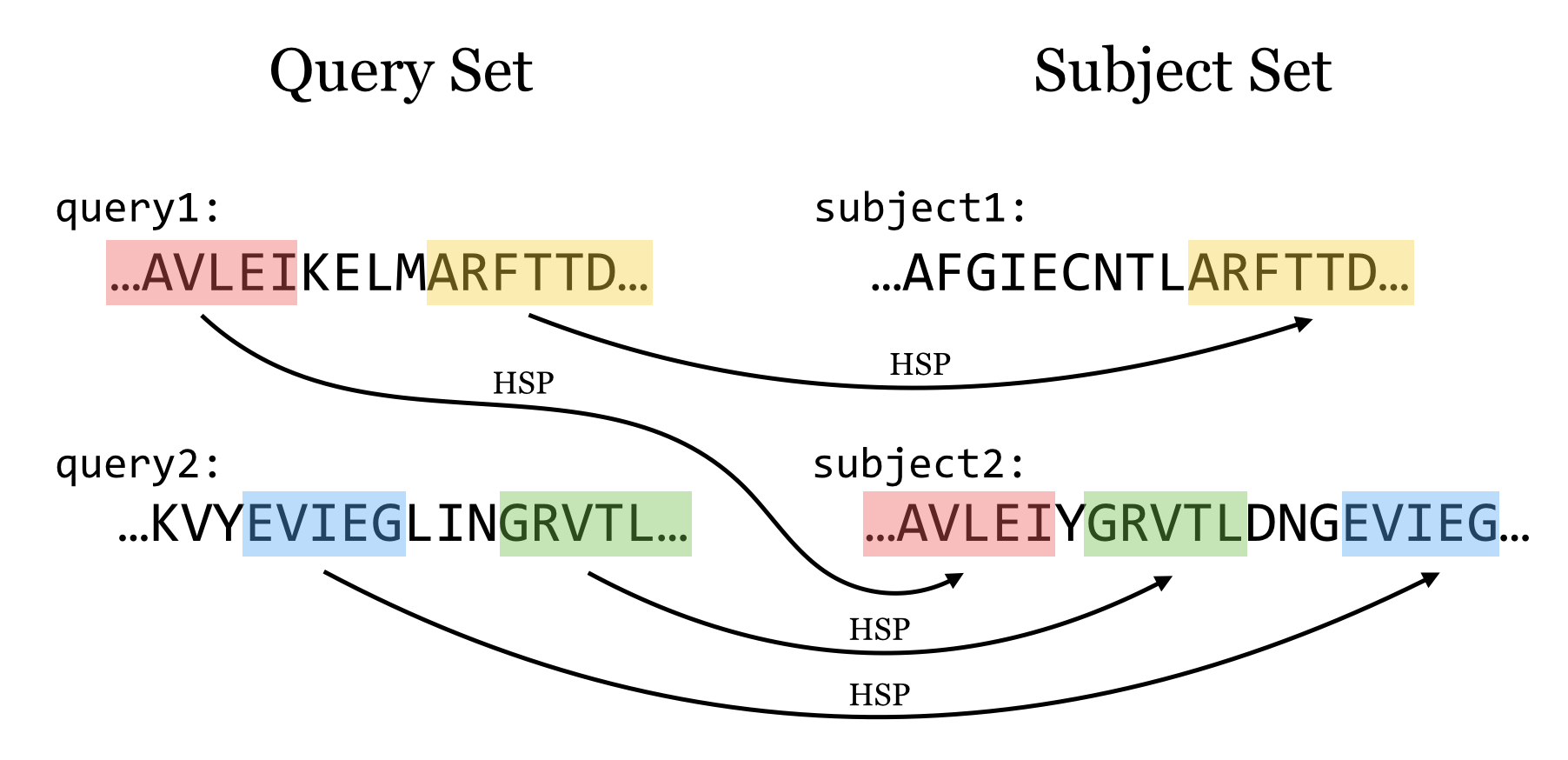
E-value
E-value = the number of HSPs having score S (or higher) expected to occur by chance.
Smaller E-value, more significant in statistics Bigger E-value , by chance
E[# occurrences of a string of length m in reference of length L] ~ L/4m
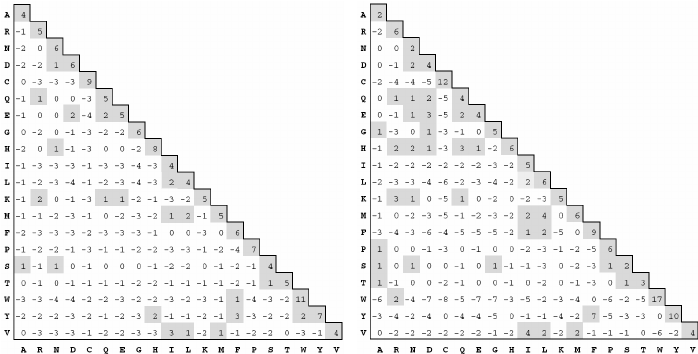
PAM and BLOSUM Matrices
Two different kinds of amino acid scoring matrices, PAM (Percent Accepted Mutation) and BLOSUM (BLOcks SUbstitution Matrix), are in wide use. The PAM matrices were created by Margaret Dayhoff and coworkers and are thus sometimes referred to as the Dayhoff matrices. These scoring matrices have a strong theoretical component and make a few evolutionary assumptions. The BLOSUM matrices, on the other hand, are more empirical and derive from a larger data set. Most researchers today prefer to use BLOSUM matrices because in silico experiments indicate that searches employing BLOSUM matrices have higher sensitivity.
There are several PAM matrices, each one with a numeric suffix. The PAM1 matrix was constructed with a set of proteins that were all 85 percent or more identical to one another. The other matrices in the PAM set were then constructed by multiplying the PAM1 matrix by itself: 100 times for the PAM100; 160 times for the PAM160; and so on, in an attempt to model the course of sequence evolution. Though highly theoretical (and somewhat suspect), it is certainly a reasonable approach. There was little protein sequence data in the 1970s when these matrices were created, so this approach was a good way to extrapolate to larger distances.
Protein databases contained many more sequences by the 1990s so a more empirical approach was possible. The BLOSUM matrices were constructed by extracting ungapped segments, or blocks, from a set of multiply aligned protein families, and then further clustering these blocks on the basis of their percent identity. The blocks used to derive the BLOSUM62 matrix, for example, all have at least 62 percent identity to some other member of the block.
BLAST has a number of possible programs to run depending on whether you have nucleotide or protein sequences:
nucleotide query and nucleotide db - blastn nucleotide query and nucleotide db - tblastx (includes six frame translation of query and db sequences) nucleotide query and protein db - blastx (includes six frame translation of query sequences) protein query and nucleotide db - tblastn (includes six frame translation of db sequences) protein query and protein db - blastp

BLAST Process



Web BLAST (NCBI)
https://blast.ncbi.nlm.nih.gov/Blast.cgi
NCBI provides a free web-based BLAST service that allows you to search your sequences against NCBI’s massive databases without installing any software. This is the easiest way to get started with BLAST.
How to Use Web BLAST
- Go to https://blast.ncbi.nlm.nih.gov/Blast.cgi
- Choose the appropriate BLAST program based on your query and database types:
| Program | Click on | Query Type | Database Type |
|---|---|---|---|
| blastn | Nucleotide BLAST | Nucleotide | Nucleotide (nt, refseq_rna, etc.) |
| blastp | Protein BLAST | Protein | Protein (nr, swissprot, pdb, etc.) |
| blastx | blastx | Nucleotide | Protein (translated query) |
| tblastn | tblastn | Protein | Nucleotide (translated database) |
| tblastx | tblastx | Nucleotide | Nucleotide (both translated) |
- Paste your sequence(s) in FASTA format or enter accession numbers in the query box
- Select the target database (e.g.,
nr,nt,swissprot,refseq_protein) - (Optional) Set organism filter, E-value threshold, or other parameters under “Algorithm parameters”
- Click BLAST and wait for results
Web BLAST Interface Overview

Input Section
- Enter Query Sequence: Paste FASTA sequence or enter accession/GI numbers
- Upload file: Upload a FASTA file from your computer
- Job Title: Name your search for later reference
- Query Subrange: Search only a portion of your query (specify From/To positions)
Choose Search Set
- Database: Select which database to search against
- nr (non-redundant): Comprehensive protein database (largest)
- nt: Comprehensive nucleotide database
- refseq_protein/refseq_rna: NCBI Reference Sequences (curated)
- swissprot: Manually reviewed UniProt entries
- pdb: Protein Data Bank sequences (for proteins with known 3D structure)
- Organism: Restrict search to specific taxonomy (e.g., “Homo sapiens”, “Viridiplantae”)
- Exclude: Exclude specific organisms or uncultured/environmental sequences
Algorithm Parameters
- Max target sequences: Number of aligned sequences to display (default: 100)
- Short queries: Automatically adjust parameters for short sequences
- Expect threshold: E-value cutoff (default: 10, use 0.001 or lower for stringent searches)
- Word size: Initial seed length
- Scoring matrix: BLOSUM62 (default), BLOSUM80, PAM250, etc.
- Gap costs: Gap open and extension penalties
- Compositional adjustments: Statistical corrections for sequence composition bias
- Filters: Low complexity filtering, species-specific repeats
Reading Web BLAST Results
The results page has several sections:
Graphic Summary
A color-coded bar diagram showing where hits align to your query. Colors indicate alignment scores:
- Red (>200): Very strong match
- Pink (80-200): Strong match
- Green (50-80): Moderate match
- Blue (40-50): Weak match
- Black (<40): Very weak match
Descriptions Table
A ranked list of database hits showing:
- Description: Name/annotation of the matching sequence
- Max Score / Total Score: Alignment score (higher = better match)
- Query Cover: Percentage of your query aligned to the subject
- E-value: Statistical significance (lower = more significant)
- Per. Ident: Percent identity across the alignment
- Accession: Database identifier
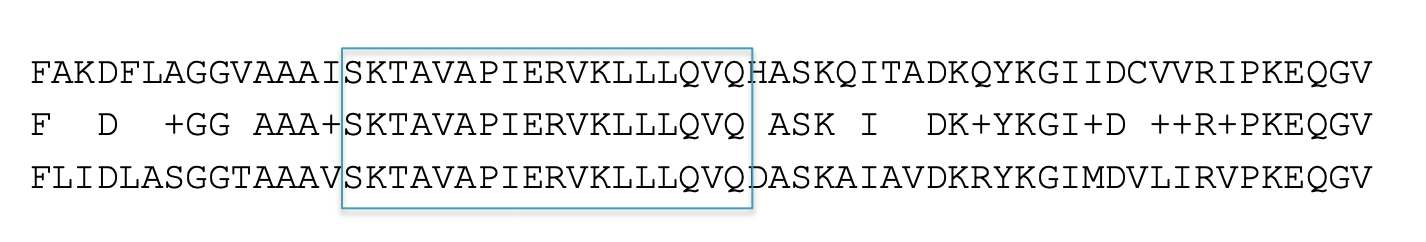
Alignments
Detailed pairwise alignments between your query and each hit, showing:
- Matched positions (letters)
- Mismatches (different letters)
- Gaps (dashes
-) - For protein: conservative substitutions (shown with
+in the middle line)
Web BLAST vs Local BLAST
| Feature | Web BLAST | Local BLAST |
|---|---|---|
| Setup required | None (just a browser) | Install BLAST+, download databases |
| Database availability | All NCBI databases (nr, nt, refseq, etc.) | Must download each database |
| Database size limit | Access to full NCBI databases (>100 GB) | Limited by local disk space |
| Query limit | ~100 sequences per submission | Unlimited |
| Speed | Depends on server load; queued | Depends on local hardware |
| Customization | Limited to web interface options | Full command-line control |
| Reproducibility | Results may change as databases update | Fixed database version |
| Output formats | HTML, XML, CSV, tabular | All formats (including custom tabular) |
| Custom databases | Not supported | Build your own with makeblastdb |
| Batch processing | Limited (use Batch Entrez for large jobs) | Full scripting/automation |
| Best for | Quick searches, small number of queries | Large-scale analysis, pipelines, custom DBs |
When to use Web BLAST: Use it for quick, one-off searches of a few sequences against NCBI databases. It requires no setup and gives you access to the latest, most comprehensive databases.
When to use Local BLAST: Use it when you have many query sequences (>100), need custom databases, require specific output formats, or need reproducible results with a fixed database version.
SmartBLAST
https://blast.ncbi.nlm.nih.gov/smartblast/
SmartBLAST is a simplified BLAST interface designed to quickly identify what a protein sequence is. It is ideal for students and researchers who need a fast answer without configuring parameters.
How SmartBLAST Works
- Paste a protein sequence (no FASTA header needed)
- SmartBLAST automatically runs the search and returns results in seconds
- Results show the top hits from landmark (well-annotated) sequences
What Makes SmartBLAST Different from Regular BLAST
| Feature | Regular Web BLAST | SmartBLAST |
|---|---|---|
| Input | FASTA format, multiple sequences | Single protein sequence (paste directly) |
| Configuration | Many parameters to set | No configuration needed |
| Database | User selects (nr, swissprot, etc.) | Automatically uses landmark/nr |
| Speed | Can take minutes | Usually returns in seconds |
| Results | Full alignments, detailed statistics | Simplified view with top hits |
| Phylogenetic context | No | Shows a quick distance tree |
| Best for | Detailed analysis | Quick “what is this protein?” |
SmartBLAST Results Include
- Top 5 matches from well-characterized landmark proteins (curated representative sequences)
- Top hit from nr (the full non-redundant database)
- A quick phylogenetic tree showing evolutionary relationships between your query and the top hits
- Pairwise alignments with percent identity and E-values
- Direct links to full BLAST results if you want more detail
SmartBLAST is the fastest way to answer “What protein is this?” Just paste a sequence and get an immediate answer with evolutionary context. It’s especially useful in the classroom for quick demonstrations.
Magic-BLAST
https://ncbi.github.io/magicblast/
Magic-BLAST is a specialized BLAST tool designed for mapping next-generation sequencing (NGS) reads (RNA-seq and DNA-seq) against a reference genome or transcriptome. It is optimized for short reads and spliced alignments, filling a niche between traditional BLAST and dedicated read mappers like HISAT2 or STAR.
Why Magic-BLAST?
Traditional BLAST was not designed for the millions of short reads produced by NGS platforms. Dedicated aligners (BWA, Bowtie2, HISAT2) are fast but use different algorithms. Magic-BLAST bridges the gap — it uses the familiar BLAST engine but optimized for:
- Short reads (Illumina 50-300 bp)
- Long reads (PacBio, Oxford Nanopore)
- Spliced alignments (for RNA-seq reads that span intron-exon junctions)
- Paired-end reads (proper handling of insert size)
Magic-BLAST vs Other Aligners
| Feature | BLAST | Magic-BLAST | HISAT2/STAR | BWA-MEM |
|---|---|---|---|---|
| Designed for | Individual sequences | NGS reads | RNA-seq reads | DNA-seq reads |
| Spliced alignment | No | Yes | Yes | No |
| Long read support | Yes | Yes | Limited | Yes |
| Speed (millions of reads) | Very slow | Moderate | Fast | Fast |
| Output format | BLAST formats | SAM/BAM | SAM/BAM | SAM/BAM |
| Database | makeblastdb | makeblastdb or SRA accession | Custom index | Custom index |
| Requires genome index | BLAST DB | BLAST DB | Specific index | Specific index |
Installation
conda create -n magicblast -c bioconda magicblast -y
conda activate magicblast
Basic Usage
## Map paired-end reads to a reference genome
magicblast -query reads_R1.fastq -query_mate reads_R2.fastq \
-db reference_genome \
-out results.sam \
-num_threads 4 \
-splice T ## Enable spliced alignment for RNA-seq
## Map reads directly from SRA accession (no download needed!)
magicblast -sra SRR1234567 \
-db reference_genome \
-out results.sam \
-num_threads 4
Key Magic-BLAST Options
| Option | Description |
|---|---|
-query / -query_mate |
Input FASTQ/FASTA files (R1 and R2 for paired-end) |
-sra |
SRA accession number (reads are streamed directly) |
-db |
BLAST database of reference genome |
-splice |
Enable spliced alignment (T/F, default: T) |
-out |
Output file (SAM format by default) |
-num_threads |
Number of CPU threads |
-outfmt |
Output format: sam (default), tabular |
-score |
Minimum alignment score |
-max_intron_length |
Maximum intron size for spliced alignments (default: 500,000) |
Magic-BLAST’s unique feature is the ability to directly stream reads from SRA accessions without downloading FASTQ files first. This saves disk space and time for exploratory analyses. However, for production RNA-seq pipelines, dedicated aligners like HISAT2 or STAR are typically faster and produce more refined spliced alignments.
IgBLAST
https://www.ncbi.nlm.nih.gov/igblast/
IgBLAST (Immunoglobulin BLAST) is a specialized BLAST tool for analyzing immunoglobulin (Ig) and T cell receptor (TCR) sequences. It is the standard tool for antibody sequence analysis, V(D)J gene assignment, and characterizing antibody repertoires.
Why IgBLAST?
Antibody and TCR genes are unique in biology — they are assembled by somatic recombination of V (Variable), D (Diversity), and J (Joining) gene segments, followed by somatic hypermutation. Standard BLAST cannot properly analyze these sequences because:
- Antibodies are composed of multiple gene segments (V, D, J) that need to be identified separately
- Somatic hypermutation introduces many point mutations, especially in complementarity-determining regions (CDRs)
- The framework regions (FRs) and CDRs need to be annotated according to established numbering schemes (Kabat, IMGT, Chothia)
IgBLAST solves all of these problems.
What IgBLAST Reports
For each antibody/TCR sequence, IgBLAST identifies:
- V gene: Best matching germline V gene segment and allele
- D gene: Best matching D gene segment (for heavy chains)
- J gene: Best matching J gene segment
- CDR1, CDR2, CDR3: Complementarity-determining region sequences
- Framework regions: FR1, FR2, FR3, FR4 sequences
- Junction analysis: Detailed V-D-J junction with N-nucleotide additions and P-nucleotides
- Percent identity to germline: Shows the level of somatic hypermutation
- Rearrangement summary: Complete V(D)J gene assignment
Web IgBLAST
- Go to https://www.ncbi.nlm.nih.gov/igblast/
- Select Organism (human, mouse, rat, etc.)
- Choose Query type: Ig (immunoglobulin) or TCR (T cell receptor)
- Select Domain system: IMGT, Kabat, or Chothia numbering
- Paste your nucleotide or protein sequence
- Click BLAST
Command-line IgBLAST
## Install
conda create -n igblast -c bioconda igblast -y
conda activate igblast
## Run IgBLAST (requires germline database setup)
igblastn -query antibody_sequences.fasta \
-germline_db_V imgt_human_V \
-germline_db_D imgt_human_D \
-germline_db_J imgt_human_J \
-organism human \
-domain_system imgt \
-auxiliary_data optional_file/human_gl.aux \
-outfmt "7 std qseq sseq" \
-out igblast_results.txt
IgBLAST vs Regular BLAST for Antibody Sequences
| Feature | Regular BLAST | IgBLAST |
|---|---|---|
| V(D)J gene identification | No | Yes (separate V, D, J assignments) |
| CDR/FR annotation | No | Yes (CDR1, CDR2, CDR3, FR1-4) |
| Junction analysis | No | Yes (N-additions, P-nucleotides) |
| Germline databases | Generic (nr/nt) | Specialized (IMGT, built-in) |
| Somatic mutation analysis | No | Yes (% identity to germline) |
| Numbering schemes | None | IMGT, Kabat, Chothia |
| Best for | General sequence search | Antibody/TCR repertoire analysis |
IgBLAST is essential for anyone working in immunology, antibody engineering, or adaptive immune repertoire sequencing (AIRR-seq). For large-scale repertoire analysis (millions of sequences), consider pipeline tools like MiXCR, IMGT/HighV-QUEST, or Change-O that build on top of IgBLAST.
BLAST 2 Sequences (bl2seq)
BLAST 2 Sequences (bl2seq) allows you to directly compare two sequences against each other without building a database. This is useful when you want to see how two specific sequences align — for example, comparing two genes, checking if two contigs overlap, or visualizing the relationship between a query and a known reference.
Web BLAST 2 Sequences
- Go to https://blast.ncbi.nlm.nih.gov/Blast.cgi
- Select any BLAST program (e.g., Nucleotide BLAST or Protein BLAST)
- Check the “Align two or more sequences” checkbox
- Paste the first sequence in the Query box
- Paste the second sequence in the Subject box
- Click BLAST

Command-line bl2seq
In BLAST+, bl2seq functionality is built into the standard BLAST programs using the -subject flag instead of -db:
## Compare two nucleotide sequences (blastn)
blastn -query sequence1.fasta \
-subject sequence2.fasta \
-out bl2seq_results.txt \
-outfmt 0
## Compare two protein sequences (blastp)
blastp -query protein1.fasta \
-subject protein2.fasta \
-out bl2seq_results.txt \
-outfmt 0
## Translated comparison: nucleotide query vs protein subject (blastx)
blastx -query gene.fasta \
-subject protein.fasta \
-out bl2seq_results.txt \
-outfmt 0
When using
-subjectinstead of-db, you do not need to runmakeblastdb— BLAST directly compares the two files. All output format options (-outfmt) work the same way.
Common Use Cases for bl2seq
| Use Case | Program | Example |
|---|---|---|
| Compare two genes from different species | blastn | Are these orthologs? How conserved are they? |
| Compare a cDNA to genomic DNA | blastn | Find exon-intron boundaries |
| Check if two contigs overlap | blastn | Genome assembly validation |
| Compare protein isoforms | blastp | What regions differ between splice variants? |
| Compare a gene to a known protein | blastx | Verify the correct reading frame |
| Compare two genomes/chromosomes | blastn | Synteny analysis, structural rearrangements |
Tabular Output for bl2seq
You can use tabular output just like regular BLAST:
## Tabular comparison with custom columns
blastn -query seq1.fasta \
-subject seq2.fasta \
-outfmt "6 qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore" \
-out comparison.txt
Dot Plot Visualization
bl2seq results can be visualized as a dot plot — a 2D plot where one sequence is on the X-axis and the other on the Y-axis. Aligned regions appear as diagonal lines:
- Diagonal lines (bottom-left to top-right): Regions aligned in the same orientation
- Anti-diagonal lines (top-left to bottom-right): Inversions
- Horizontal/vertical offsets: Insertions or deletions
- Parallel diagonals: Duplicated regions
Tools for dot plot visualization:
- Gepard: https://cube.univie.ac.at/gepard
- D-GENIES: https://dgenies.toulouse.inra.fr/
- MUMmer/mummerplot: Command-line tool for large genome comparisons
- NCBI Web BLAST: Shows a simplified graphic alignment
## Using MUMmer for dot plot (alternative to bl2seq for large sequences)
conda install -c bioconda mummer -y
nucmer -p comparison genome1.fasta genome2.fasta
mummerplot -p dotplot --png comparison.delta
bl2seq is the simplest way to compare two sequences. For comparing entire genomes or very large sequences, consider MUMmer (nucmer/promer) which is optimized for whole-genome alignment and produces better dot plots.
Other Specialized BLAST Variants
NCBI and the community have developed several other BLAST variants for specific use cases:
PSI-BLAST (Position-Specific Iterated BLAST)
PSI-BLAST is built into the standard BLAST+ suite. It performs iterative protein searches to detect distant homologs that regular BLASTP might miss.
How it works:
- Run a standard BLASTP search
- Build a PSSM (Position-Specific Scoring Matrix) from the significant hits
- Search the database again using the PSSM instead of a generic BLOSUM matrix
- Repeat steps 2-3 for multiple iterations, detecting progressively more distant homologs
## Run PSI-BLAST with 3 iterations
psiblast -query protein.fasta \
-db nr \
-num_iterations 3 \
-evalue 0.001 \
-out psiblast_results.txt \
-out_pssm pssm_checkpoint.pssm \
-num_threads 4
PSI-BLAST is powerful for detecting remote homologs (< 20% identity) but beware of profile corruption: if a false positive enters the PSSM in early iterations, subsequent iterations will find more unrelated sequences. Always review results carefully and use strict E-value cutoffs.
DELTA-BLAST (Domain Enhanced Lookup Time Accelerated BLAST)
DELTA-BLAST searches a pre-constructed database of PSSMs (the Conserved Domain Database, CDD) before searching the protein database. This gives PSI-BLAST-level sensitivity in a single pass without iterative searching.
## Run DELTA-BLAST
deltablast -query protein.fasta \
-db nr \
-rpsdb cdd_delta \
-evalue 0.001 \
-out deltablast_results.txt \
-num_threads 4
BLAST+ Remote Search
You can search NCBI databases remotely from the command line without downloading them:
## Remote BLASTP against nr (runs on NCBI servers)
blastp -query protein.fasta -db nr -remote -out remote_results.txt -outfmt 6
## Remote BLASTN against nt
blastn -query nucleotide.fasta -db nt -remote -out remote_results.txt -outfmt 6
Remote BLAST is useful for one-off searches against databases too large to download (nr is >300 GB). However, it depends on network speed and NCBI server load, and is subject to usage limits.
RPSBlast (Reverse Position-Specific BLAST)
RPSBlast searches a protein query against a database of PSSMs (profiles), typically the Conserved Domain Database (CDD). This is the engine behind NCBI’s CD-Search (Conserved Domain Search).
## Search for conserved domains in a protein
rpsblast -query protein.fasta \
-db Cdd \
-evalue 0.01 \
-out domain_results.txt \
-outfmt "6 qseqid sseqid pident evalue bitscore stitle"
Comparison of All BLAST Variants
| Tool | Type | Best For | Sensitivity | Speed |
|---|---|---|---|---|
| blastn | Nucleotide vs nucleotide | DNA/RNA sequence search | Standard | Fast |
| blastp | Protein vs protein | Protein sequence search | Standard | Moderate |
| blastx | Translated nt vs protein | Gene finding, annotation | Standard | Slow |
| tblastn | Protein vs translated nt | Finding genes in genomes | Standard | Slow |
| tblastx | Translated nt vs translated nt | Comparing unannotated genomes | Standard | Very slow |
| PSI-BLAST | Iterative protein search | Remote homolog detection | Very high | Slow (iterative) |
| DELTA-BLAST | Domain-enhanced protein search | Remote homologs (single pass) | Very high | Moderate |
| RPSBlast | Protein vs domain profiles | Conserved domain identification | High | Fast |
| SmartBLAST | Quick protein identification | “What is this protein?” | Standard | Very fast |
| Magic-BLAST | NGS read mapping | RNA-seq, read alignment | Moderate | Moderate |
| IgBLAST | Antibody/TCR analysis | V(D)J assignment, repertoire | Specialized | Moderate |
| DIAMOND | Ultra-fast protein aligner | Large-scale blastx/blastp | Adjustable | 100-20,000x faster |
NCBI BLAST API (for programmatic access)
For automated searches beyond the web interface, NCBI provides a REST API:
## Submit a BLAST search via command line using NCBI API
## Step 1: Submit the query (returns a Request ID / RID)
curl -X POST "https://blast.ncbi.nlm.nih.gov/blast/Blast.cgi" \
-d "CMD=Put&PROGRAM=blastp&DATABASE=nr&QUERY=MASGPGGWLGPAFALRLLLAAVLQPVSAFRA"
## Step 2: Check status (replace RID with the actual Request ID)
curl "https://blast.ncbi.nlm.nih.gov/blast/Blast.cgi?CMD=Get&FORMAT_TYPE=Text&RID=YOUR_RID"
For large-scale automated BLAST searches against NCBI databases, consider using the NCBI BLAST+
remoteoption:blastp -query input.fasta -db nr -remote. This runs the search on NCBI’s servers using the local command-line interface.
UniProt
https://www.uniprot.org/
UniProt (Universal Protein Resource) is the most comprehensive and widely used protein sequence and functional annotation database. It consists of three main components:
- UniProtKB/Swiss-Prot – Manually reviewed and annotated protein entries. High quality but smaller (~570,000 entries). Each entry is curated by experts with experimental evidence and literature references.
- UniProtKB/TrEMBL – Automatically annotated protein entries that have not yet been manually reviewed. Much larger (~250 million entries). Generated from translated coding sequences in EMBL-Bank/GenBank/DDBJ nucleotide databases.
- UniRef – Clustered sets of sequences from UniProtKB to reduce redundancy. UniRef100 combines identical sequences, UniRef90 clusters at 90% identity, and UniRef50 at 50% identity. Useful for faster BLAST searches with reduced database size.
- UniParc – A comprehensive, non-redundant archive of all protein sequences from major databases. No functional annotation; serves as a sequence repository.
Why use UniProt for BLAST?
| Feature | NCBI nr | UniProtKB/Swiss-Prot | UniProtKB/TrEMBL |
|---|---|---|---|
| Size | Very large (~600M seqs) | Small (~570K seqs) | Large (~250M seqs) |
| Annotation quality | Variable | Manually curated | Automatic |
| Redundancy | Low | No redundancy | Low |
| Search speed | Slow | Very fast | Moderate |
| Best for | Comprehensive search | High-confidence annotation | Broad coverage with annotation |
- Use Swiss-Prot when you want high-confidence functional annotations and fast searches.
- Use TrEMBL (or full UniProtKB) when you need broader coverage and are willing to accept computationally predicted annotations.
- Use nr when you want the most comprehensive search across all known proteins.
Running BLASTP against UniProt locally
You can download UniProt databases for local BLAST searches:
cd ~/bch709/BLAST
## Download Swiss-Prot (small, manually curated)
wget https://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/complete/uniprot_sprot.fasta.gz
gunzip uniprot_sprot.fasta.gz
makeblastdb -in uniprot_sprot.fasta -dbtype prot -parse_seqids
## Download individual protein FASTA files from UniProt REST API
wget https://rest.uniprot.org/uniprotkb/P12345.fasta
wget https://rest.uniprot.org/uniprotkb/P01308.fasta
## Run BLASTP against Swiss-Prot (using human insulin, P01308, as the query)
blastp -query P01308.fasta \
-db uniprot_sprot.fasta \
-outfmt "6 qseqid sseqid pident evalue bitscore stitle" \
-evalue 1e-05 \
-max_target_seqs 5 \
-num_threads 4 \
-out blastp_swissprot.txt
Swiss-Prot entries use accession numbers (e.g.,
P12345) and entry names (e.g.,INS_HUMANfor human insulin). Thestitlecolumn in BLAST output will show the full description line including gene name, organism, and protein function.Example entries on UniProt:
BLASTN example
Run blastn against the nt database.
ATGAAAGCGAAGGTTAGCCGTGGTGGCGGTTTTCGCGGTGCGCTGAACTA
CGTTTTTGACGTTGGCAAGGAAGCCACGCACACGAAAAACGCGGAGCGAG
TCGGCGGCAACATGGCCGGGAATGACCCCCGCGAACTGTCGCGGGAGTTC
TCAGCCGTGCGCCAGTTGCGCCCGGACATCGGCAAGCCCGTCTGGCATTG
CTCGCTGTCACTGCCTCCCGGCGAGCGCCTGAGCGCCGAGAAGTGGGAAG
CCGTCGCGGCTGACTTCATGCAGCGCATGGGCTTTGACCAGACCAATACG
CCGTGGGTGGCCGTGCGCCACCAGGACACGGACAAGGATCACATCCACAT
CGTGGCCAGCCGGGTAGGGCTGGACGGGAAAGTGTGGCTGGGCCAGTGGG
AAGCCCGCCGCGCCATCGAGGCGACCCAAGAGCTTGAGCATACCCACGGC
CTGACCCTGACGCCGGGGCTGGGCGATGCGCGGGCCGAGCGCCGGAAGCT
GACCGACAAGGAGATCAACATGGCCGTGAGAACGGGCGATGAACCGCCGC
GCCAGCGTCTGCAACGGCTGCTGGATGAGGCGGTGAAGGACAAGCCGACC
GCGCTAGAACTGGCCGAGCGGCTACAGGCCGCAGGCGTAGGCGTCCGGGC
AAACCTCGCCAGCACCGGGCGCATGAACGGCTTTTCCTTCGAGGTGGCCG
GAGTGCCGTTCAAAGGCAGCGACTTGGGCAAGGGCTACACATGGGCGGGG
CTACAGAAAGCAGGGGTGACTTATGACGAAGCTAGAGACCGTGCGGGCCT
TGAACGATTCAGGCCCACAGTTGCAGATCGTGGAGAGCGTCAGGACGTTG
CAGCAGTCCGTGAGCCTGATGCACGAGGACTTGAAGCGCCTACCGGGCGC
AGTCTCGACCGAGACGGCGCAGACCTTGGAACCGCTGGCCCGACTCCGGC
AGGACGTGACGCAGGTTCTGGAAGCCTACGACAAGGTGACGGCCATTCAG
CGCAAGACGCTGGACGAGCTGACGCAGCAGATGAGCGCGAGCGCGGCGCA
GGCCTTCGAGCAGAAGGCCGGGAAGCTGGACGCGACCATCTCCGACCTGT
CGCGCAGCCTGTCAGGGCTGAAAACGAGCCTCAGCAGCATGGAGCAGACC
GCGCAGCAGGTGGCGACCTTGCCGGGCAAGCTGGCGAGCGCACAGCAGGG
CATGACGAAAGCCGCCGACCAACTGACCGAGGCAGCGAACGAGACGCGCC
CGCGCCTTTGGCGGCAGGCGCTGGGGCTGATTCTGGCCGGGGCCGTGGGC
GCGATGCTGGTAGCGACTGGGCAAGTCGCTTTAAACAGGCTAGTGCCGCC
AAGCGACGTGCAGCAGACGGCAGACTGGGCCAACGCGATTTGGAACAAGG
CCACGCCCACGGAGCGCGAGTTGCTGAAACAGATCGCCAATCGGCCCGCG
AACTAGACCCGACCGCCTACCTTGAGGCCAGCGGCTACACCGTGAAGCGA
GAAGGGCGGCACCTGTCCGTCAGGGCGGGCGGTGATGAGGCGTACCGCGT
GACCCGGCAGCAGGACGGGCGCTGGCTCTGGTGCGACCGCTACGGCAACG
ACGGCGGGGACAATATCGACCTGGTGCGCGAGATCGAACCCGGCACCGGC
TACGCCGAGGCCGTCTATCGGCTTTCAGGTGCGCCGACAGTCCGGCAGCA
ACCGCGCCCGAGCGAGCCGAAGCGCCAACCGCCGCAGCTACCGGCGCAAG
GGCTGGCAGCCCGCGAGCATGGCCGCGACTACCTCAAGGGCCGGGGCATC
AGCCAGGACACCATCGAGCACGCCGAGAAGGCGGGCATGGTGCGCTATGC
AGACGGTGGAGTGCTGTTCGTCGGCTACGACCGTGCAGGCACCGCGCAGA
ACGCCACACGCCGCGCCATTGCCCCCGCTGACCCGGTGCAGAAGCGCGAC
CTACGCGGCAGCGACAAGAGCTATCCGCCGATCCTGCCGGGCGACCCGGC
AAAGGTCTGGATCGTGGAAGGTGGCCCGGATGCGCTGGCCCTGCACGACA
TCGCCAAGCGCAGCGGCCAGCAGCCGCCCACCGTCATCGTGTCAGGCGGG
GCGAACGTGCGCAGCTTCTTGGAGCGGGCCGACGTGCAAGCGATCCTGAA
GCGGGCCGAGCGCGTCACCGTGGCCGGGGAAAACGAGAAGAACCCCGAGG
CGCAGGCAAAGGCCGACGCCGGGCACCAGAAGCAGGCGCAGCGGGTGGCC
AAAATCACCGGGCGCGAGGTGCGCCAATGGACGCCGAAGCCCGAGCACGG
CAAGGACTTGGCCGACATGAACGCCCGGCAGGTGGCAGAGATCGAGCGCA
AGCGACAGGCCGAGATCGAGGCCGAAAGAGCACGAAACCGCGAGCTTTCA
CGCAAGAGCCGGAGGTATGATGGCCCCAGCTTCGGCAGATAA
BLASTP Query
Do a BLASTP on NCBI website with the following protein against nr, but limit the organism to cetartiodactyla using default parameters:
MASGPGGWLGPAFALRLLLAAVLQPVSAFRAEFSSESCRELGFSSNLLCSSCDLLGQFSL
LQLDPDCRGCCQEEAQFETKKYVRGSDPVLKLLDDNGNIAEELSILKWNTDSVEEFLSEK
LERI
Have a look at the multiple sequence alignment, can you explain the results?
Do a similar blastp vs UniProtKB (UniProt) without post filtering.
Running a standalone BLAST program
Location
cd ~/bch709/BLAST
ENV
conda activate blast
Running a standalone BLAST program
Create the index for the target database using makeblastdb; Choose the task program: blastn, blastp, blastx, tblatx, psiblast or deltablast; Set the configuration for match, mismatch, gap-open penalty, gap-extension penalty or scoring matrix; Set the word size; Set the E-value threshold; Set the output format and the number of output results
Standalone BLAST
In addition to providing BLAST sequence alignment services on the web, NCBI also makes these sequence alignment utilities available for download through FTP. This allows BLAST searches to be performed on local platforms against databases downloaded from NCBI or created locally. These utilities run through DOS-like command windows and accept input through text-based command line switches. There is no graphic user interface
https://www.ncbi.nlm.nih.gov/books/NBK52640/
https://ftp.ncbi.nlm.nih.gov/blast/db/
NR vs NT
At NCBI they are two different things as well. ‘nr’ is a database of protein sequences and ‘nt’ is nucleotide. At one time ‘nr’ meant non-redundant but it stopped being non-redundant a while ago. nt is a nucleotide database, while nr is a protein database (in amino acids)
Standalone BLAST
- Download the database.
- Use makeblastdb to build the index.
- Change the scoring matrix, record the changes in the alignment results and interpret the results.
Download Database
cd ~/bch709/BLAST
wget https://ftp.ncbi.nlm.nih.gov/refseq/release/plant/plant.1.protein.faa.gz
Expected output (truncated):
Resolving ftp.ncbi.nlm.nih.gov ... 130.14.250.7
Connecting to ftp.ncbi.nlm.nih.gov:443 ... connected.
HTTP request sent, awaiting response... 200 OK
Length: 24342023 (23M) [application/x-gzip]
Saving to: 'plant.1.protein.faa.gz'
How many sequences in plant.1.protein.faa.gz
seqkit stats plant.1.protein.faa.gz
Download Input file
Download the Arabidopsis thaliana TAIR10 CDS sequences:
cd ~/bch709/BLAST
wget https://ftp.ensemblgenomes.org/pub/plants/release-60/fasta/arabidopsis_thaliana/cds/Arabidopsis_thaliana.TAIR10.cds.all.fa.gz -O Athaliana_TAIR10.cds.fa.gz --no-check-certificate
Example Input sequence
seqkit stats Athaliana_TAIR10.cds.fa.gz -T
Expected output (values may vary slightly depending on the source version; sample below is from Ensembl Plants release-60):
file format type num_seqs sum_len min_len avg_len max_len
Athaliana_TAIR10.cds.fa.gz FASTA DNA 48321 62644401 3 1296.4 16203
Subsampling by SeqKit
FASTA and FASTQ are basic and ubiquitous formats for storing nucleotide and protein sequences. Common manipulations of FASTA/Q file include converting, searching, filtering, deduplication, splitting, shuffling, and sampling. Existing tools only implement some of these manipulations, and not particularly efficiently, and some are only available for certain operating systems. Furthermore, the complicated installation process of required packages and running environments can render these programs less user friendly.
This project describes a cross-platform ultrafast comprehensive toolkit for FASTA/Q processing. SeqKit provides executable binary files for all major operating systems, including Windows, Linux, and Mac OS X, and can be directly used without any dependencies or pre-configurations. SeqKit demonstrates competitive performance in execution time and memory usage compared to similar tools. The efficiency and usability of SeqKit enable researchers to rapidly accomplish common FASTA/Q file manipulations.
Run BLAST
Make BLAST DB
makeblastdb -in your-nucleotide-db.fa -dbtype nucl ###for nucleotide sequence
makeblastdb -in your-protein-db.fas -dbtype prot ###for protein sequence
Run BLASTX
cd ~/bch709/BLAST
gunzip plant.1.protein.faa.gz
makeblastdb -in plant.1.protein.faa -dbtype prot -parse_seqids
seqkit sample -n 100 Athaliana_TAIR10.cds.fa.gz > ATH_100.fasta
blastx -query ATH_100.fasta -db plant.1.protein.faa -outfmt 6 -evalue 1e-10 -max_target_seqs 1 -num_threads 4 -out blastx_results.txt
head -3 blastx_results.txt
The
-parse_seqidsflag stores the original sequence IDs in the BLAST database, which is required for laterblastdbcmd -entry_batchlookups (e.g. in the Reciprocal Best Hit workflow below). Without itblastdbcmdskips every accession and returns an empty FASTA file.
Expected output (sample first 3 hits, will vary because seqkit sample is randomized):
AT5G61960.5 XP_023635731.1 91.529 909 66 4 4 2724 9 908 0.0 1477
AT2G30460.1 XP_024004202.1 94.302 351 20 0 1 1053 1 351 0.0 597
AT4G10850.1 XP_023636862.1 84.646 254 39 0 1 762 1 254 2.82e-162 451
Tab output (default -outfmt 6 columns)
qseqid Query sequence ID
sseqid Subject (ie DB) sequence ID
pident Percent Identity across the alignment
length Alignment length
mismatch # of mismatches
gapopen Number of gap openings
qstart Start of alignment in query
qend End of alignment in query
sstart Start of alignment in subject
send End of alignment in subject
evalue E-value
bitscore Bit score
BLAST Key Options Explained
-outfmt : Output Format
Controls how results are displayed. This is one of the most important BLAST options.
| Value | Format | Description |
|---|---|---|
| 0 | Pairwise | Default human-readable alignment output |
| 1 | Query-anchored with identities | Shows alignment relative to query |
| 2 | Query-anchored without identities | Same as 1 but without identity markers |
| 3 | Flat query-anchored with identities | Flat version of format 1 |
| 4 | Flat query-anchored without identities | Flat version of format 2 |
| 5 | BLAST XML | Machine-readable XML format |
| 6 | Tabular | Tab-separated, no headers (most commonly used for parsing) |
| 7 | Tabular with comments | Same as 6 but includes header lines starting with # |
| 8 | Seqalign (text ASN.1) | Text ASN.1 format |
| 10 | CSV | Comma-separated values |
| 11 | BLAST archive (ASN.1) | Can be converted to other formats later with blast_formatter |
| 12 | Seqalign (JSON) | JSON format |
| 13 | Multiple-file JSON | JSON output split per query |
| 14 | Multiple-file XML2 | XML2 output split per query |
| 15 | Single-file JSON | Single JSON file |
| 16 | Single-file XML2 | Single XML2 file |
| 17 | SAM | Sequence Alignment/Map format (useful for genomics pipelines) |
| 18 | Organism Report | Summary report organized by organism |
Custom tabular columns (used with -outfmt 6 or -outfmt 7):
blastx -query ATH_100.fasta -db plant.1.protein.faa \
-outfmt "6 qseqid sseqid pident length evalue bitscore stitle"
All available column specifiers:
| Specifier | Description |
|---|---|
qseqid |
Query sequence ID |
sseqid |
Subject sequence ID |
pident |
Percentage of identical matches |
length |
Alignment length |
mismatch |
Number of mismatches |
gapopen |
Number of gap openings |
qstart / qend |
Start/end of alignment in query |
sstart / send |
Start/end of alignment in subject |
evalue |
Expect value |
bitscore |
Bit score |
score |
Raw score |
qlen |
Query sequence length |
slen |
Subject sequence length |
qcovs |
Query coverage per subject |
qcovhsp |
Query coverage per HSP |
stitle |
Subject title (description) |
salltitles |
All subject titles |
sallseqid |
All subject sequence IDs |
qframe |
Query frame |
sframe |
Subject frame |
nident |
Number of identical matches |
positive |
Number of positive-scoring matches |
gaps |
Total number of gaps |
ppos |
Percentage of positive-scoring matches |
staxids |
Subject taxonomy ID(s) |
sscinames |
Subject scientific name(s) |
scomnames |
Subject common name(s) |
-evalue : E-value Threshold
The E-value (Expect value) represents the number of alignments with a given score that would be expected by chance in a database of that size. It is the most important statistical measure in BLAST.
-evalue 1e-05 ## Only report hits with E-value <= 0.00001
| E-value | Interpretation |
|---|---|
| < 1e-50 | Nearly identical sequences |
| 1e-50 to 1e-10 | Very strong homology |
| 1e-10 to 1e-05 | Strong homology, likely related |
| 1e-05 to 0.01 | Moderate homology, possible relationship |
| 0.01 to 1 | Weak hit, borderline significance |
| > 1 | Not significant, likely random match |
| 10 (default) | BLAST default; includes many weak/random hits |
The E-value depends on database size. The same alignment will have a lower (more significant) E-value in a smaller database. When comparing results across different databases, consider this effect.
-num_threads : Number of CPU Threads
Controls parallelism for faster searches. More threads = faster search, up to a limit.
-num_threads 4 ## Use 4 CPU cores
Check how many cores are available:
nproc ## Linux
sysctl -n hw.ncpu ## macOS
BLAST scales well up to ~8 threads. Beyond that, diminishing returns are common. If you have a very large query file, splitting the input (e.g., with DCBLAST or
seqkit split) and running separate jobs is more efficient than using many threads on a single job.
-max_target_seqs : Maximum Number of Target Sequences
Limits the number of subject sequences reported for each query.
-max_target_seqs 5 ## Report up to 5 hits per query
Important:
-max_target_seqsdoes NOT guarantee the top N hits by E-value. BLAST uses a heuristic search and keeps the first N qualifying hits encountered. To reliably get the best N hits, set-max_target_seqsto a higher number and then sort the output by E-value afterward.
-out : Output File
By default, BLAST prints results to the terminal (stdout). Use -out to save to a file:
-out blastx_results.txt
-word_size : Word Size
Controls the initial seed length for finding matches. Smaller values increase sensitivity but decrease speed.
| Program | Default | Sensitive | Fast |
|---|---|---|---|
| blastn | 11 | 7 | 28 |
| blastp | 3 | 2 | 6 |
| blastx | 3 | 2 | 6 |
-word_size 7 ## More sensitive nucleotide search
-matrix : Scoring Matrix (protein BLAST only)
Specifies the substitution matrix for scoring amino acid alignments.
| Matrix | Best for | Identity Range |
|---|---|---|
| BLOSUM62 | General purpose (default) | 30-40% identity |
| BLOSUM80 | Closely related sequences | >50% identity |
| BLOSUM45 | Distantly related sequences | <30% identity |
| PAM250 | Distantly related sequences | <30% identity |
| PAM30 | Closely related sequences | >60% identity |
-matrix BLOSUM80 ## For closely related protein sequences
-gapopen / -gapextend : Gap Penalties
Control the cost of introducing and extending gaps in alignments.
| Matrix | Default gapopen | Default gapextend |
|---|---|---|
| BLOSUM62 | 11 | 1 |
| BLOSUM80 | 10 | 1 |
| BLOSUM45 | 15 | 2 |
-gapopen 11 -gapextend 1 ## Default for BLOSUM62
-query_cov and -subject_cov : Coverage Filters
Filter hits by minimum query or subject coverage percentage (BLAST+ 2.12+):
-qcov_hsp_perc 70 ## Only report HSPs covering at least 70% of the query
-max_hsps : Maximum HSPs per Subject
Limits the number of HSPs (High-Scoring Segment Pairs) reported per query-subject pair:
-max_hsps 1 ## Report only the best HSP for each query-subject pair
Putting It All Together
blastx -query ATH_100.fasta \
-db plant.1.protein.faa \
-out blastx_results.txt \
-outfmt "6 qseqid sseqid pident length evalue bitscore qcovs stitle" \
-evalue 1e-10 \
-max_target_seqs 5 \
-num_threads 4 \
-matrix BLOSUM62 \
-word_size 3 \
-max_hsps 1 \
-qcov_hsp_perc 50
This command:
- Translates nucleotide query in all 6 frames and searches against protein DB
- Saves tabular output with custom columns including query coverage and subject description
- Only reports hits with E-value <= 1e-10
- Reports up to 5 target sequences per query
- Uses 4 CPU threads
- Uses BLOSUM62 scoring matrix
- Only keeps HSPs covering at least 50% of the query
- Reports only the best HSP per query-subject pair
Question
- find the option below within BLASTX
- Set output to file
- Set tabular output format
- Set maximum target sequence to one
- Set threads (CPU) to 32
- Set evalue threshold to 1e-30
Answer
blastx -query ATH_100.fasta -db plant.1.protein.faa \ -out output.txt \ -outfmt 6 \ -max_target_seqs 1 \ -num_threads 32 \ -evalue 1e-30
DCBLAST
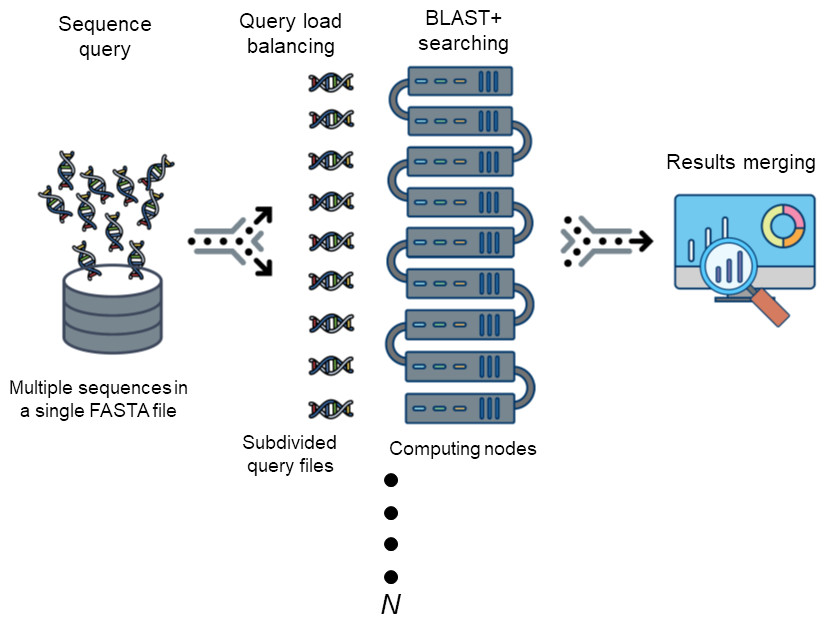
The Basic Local Alignment Search Tool (BLAST) is by far best the most widely used tool in for sequence analysis for rapid sequence similarity searching among nucleic acid or amino acid sequences. Recently, cluster, HPC, grid, and cloud environmentshave been are increasing more widely used and more accessible as high-performance computing systems. Divide and Conquer BLAST (DCBLAST) has been designed to perform run on grid system with query splicing which can run National Center for Biotechnology Information (NCBI) BLASTBLAST search comparisons over withinthe cluster, grid, and cloud computing grid environment by using a query sequence distribution approach NCBI BLAST. This is a promising tool to accelerate BLAST job dramatically accelerates the execution of BLAST query searches using a simple, accessible, robust, and practical approach.
- DCBLAST can run BLAST job across HPC.
- DCBLAST suppport all NCBI-BLAST+ suite.
- DCBLAST generate exact same NCBI-BLAST+ result.
- DCBLAST can use all options in NCBI-BLAST+ suite.
Requirement
Following basic softwares are needed to run
- Perl (Any version 5+)
which perl
perl --version
- NCBI-BLAST+ (Any version) for easy approach, you can download binary version of blast from below link. https://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/
For using recent version, please update BLAST path in config.ini
which blastn
- Sun Grid Engine (Any version)
which qsub - Slurm
which sbatch - Grid cloud or distributed computing system.
Prerequisites
The following Perl modules are required:
- Path::Tiny
- Data::Dumper
- Config::Tiny
Install prerequisites with the following command:
cpan `cat requirement`
or
cpanm `cat requirement`
or
cpanm Path::Tiny Data::Dumper Config::Tiny
We strongly recommend to use Perlbrew http://perlbrew.pl/ to avoid having to type sudo
We also recommend to use ‘cpanm’ https://github.com/miyagawa/cpanminus
Prerequisites by Conda
conda activate blast
conda install -c bioconda perl-path-tiny blast perl-data-dumper perl-config-tiny -y
Installation
The program is a single file Perl scripts. Copy it into executive directories.
We recommend to copy it on scratch disk.
cd ~/bch709/BLAST
## If you haven't already downloaded and unzipped the input file:
# wget https://ftp.ensemblgenomes.org/pub/plants/release-60/fasta/arabidopsis_thaliana/cds/Arabidopsis_thaliana.TAIR10.cds.all.fa.gz -O Athaliana_TAIR10.cds.fa.gz
# gunzip Athaliana_TAIR10.cds.fa.gz
git clone https://github.com/wyim-pgl/DCBLAST.git
cd DCBLAST/DCBLAST-SLURM
pwd
chmod 775 dcblast.pl
perl dcblast.pl
Help
Usage : dcblast.pl --ini config.ini --input input-fasta --size size-of-group --output output-filename-prefix --blast blast-program-name
--ini <ini filename> ##config file ex)config.ini
--input <input filename> ##query fasta file
--size <output size> ## size of chunks usually all core x 2, if you have 160 core all nodes, you can use 320. please check it to your admin.
--output <output filename> ##output folder name
--blast <blast name> ##blastp, blastx, blastn and etcs.
--dryrun Option will only split fasta file into chunks
Configuration
Please edit config.ini with nano before you run!!
[dcblast]
##Name of job (will use for SGE job submission name)
job_name_prefix=dcblast
[blast]
##BLAST options
##BLAST path (your blast+ path); run "which blastn" then remove "blastn" from the path
path=~/miniconda3/envs/blast/bin/
##DB path (build your own BLAST DB)
##example
##makeblastdb -in example/test_db.fas -dbtype nucl (for nucleotide sequence)
##makeblastdb -in example/your-protein-db.fas -dbtype prot (for protein sequence)
db=~/bch709/BLAST/plant.1.protein.faa
##Evalue cut-off (See BLAST manual)
evalue=1e-05
##number of threads in each job. If your CPU is AMD it needs to be set 1.
num_threads=2
##Max target sequence output (See BLAST manual)
max_target_seqs=10
##Output format (See BLAST manual)
outfmt=6
##any other option can be add it this area
#matrix=BLOSUM62
#gapopen=11
#gapextend=1
[oldsge]
##Grid job submission commands (for SGE-based HPC systems)
pe=SharedMem 1
M=your@email
q=common.q
j=yes
o=log
cwd=
[slurm]
##Slurm job submission settings (adjust for your cluster)
time=04:00:00
cpus-per-task=1
mem-per-cpu=800M
ntasks=1
output=log
error=error
partition=cpu-core-0
account=cpu-s5-bch709-3
mail-type=all
mail-user=your@email
If you need any other options for your enviroment please contant us or admin
PBS & LSF need simple code hack. If you need it please request through issue.
Run DCBLAST
Run (–dryrun option will only split fasta file into chunks)
perl dcblast.pl --ini config.ini --input ~/bch709/BLAST/Athaliana_TAIR10.cds.fa --output test --size 100 --blast blastx
Check job status:
squeue ## for Slurm
or
qstat ## for SGE
It usually finishes within up to 20 min depending on HPC status and CPU speed.
Running BLAST Locally Without a Job Scheduler
If you do not have access to an HPC cluster (Slurm/SGE), you can run BLAST directly on your local machine:
cd ~/bch709/BLAST
## Make sure your database is built
makeblastdb -in plant.1.protein.faa -dbtype prot -parse_seqids
## Run blastx locally using multiple threads
blastx -query Athaliana_TAIR10.cds.fa \
-db plant.1.protein.faa \
-out blastx_results.txt \
-outfmt 6 \
-evalue 1e-05 \
-max_target_seqs 10 \
-num_threads 4
For local runs, adjust
-num_threadsto the number of CPU cores available on your machine. You can check withnproc(Linux) orsysctl -n hw.ncpu(macOS).
Citation
Won C. Yim and John C. Cushman (2017) Divide and Conquer BLAST: using grid engines to accelerate BLAST and other sequence analysis tools. PeerJ 10.7717/peerj.3486 https://peerj.com/articles/3486/
BLAST Output Formats
BLAST supports multiple output formats controlled by the -outfmt option. Understanding these is essential for downstream analysis.
| Format | Description |
|---|---|
| 0 | Pairwise (default, human-readable) |
| 5 | BLAST XML |
| 6 | Tabular (without headers) |
| 7 | Tabular (with comment headers) |
| 8 | Text ASN.1 |
| 10 | CSV (comma-separated values) |
| 11 | BLAST archive (ASN.1) |
Custom Tabular Output
You can customize which columns appear in tabular output (-outfmt 6 or -outfmt 7) by specifying field names:
blastx -query ATH_100.fasta \
-db plant.1.protein.faa \
-outfmt "6 qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore qcovs stitle" \
-evalue 1e-05 \
-max_target_seqs 1 \
-num_threads 4 \
-out blastx_custom.txt
Additional useful columns:
qcovs- Query coverage per subjectstitle- Subject title (description line)slen- Subject sequence lengthqlen- Query sequence lengthqcovhsp- Query coverage per HSP
Filtering and Parsing BLAST Results
Using awk to filter results
## Filter for hits with >80% identity and e-value < 1e-10
awk '$3 > 80 && $11 < 1e-10' blastx_results.txt
## Count number of unique query sequences with hits
awk '{print $1}' blastx_results.txt | sort -u | wc -l
## Get the best hit (lowest e-value) for each query
sort -k1,1 -k11,11g blastx_results.txt | sort -k1,1 -u
Using SeqKit to extract sequences
## Extract query IDs from BLAST results
awk '{print $1}' blastx_results.txt | sort -u > hit_ids.txt
## Extract FASTA sequences for those IDs
seqkit grep -f hit_ids.txt Athaliana_TAIR10.cds.fa > hits.fasta
Reciprocal Best BLAST Hit (RBH)
Reciprocal Best BLAST Hit is a common method to identify putative orthologs between two species. The logic is simple: if gene A in species 1 finds gene B as its best hit in species 2, and gene B finds gene A as its best hit in species 1, then A and B are reciprocal best hits and likely orthologs.
Step 1: Forward BLAST (Species 1 vs Species 2)
cd ~/bch709/BLAST
## Download a second protein database (e.g., rice)
wget https://ftp.ncbi.nlm.nih.gov/refseq/release/plant/plant.2.protein.faa.gz
gunzip plant.2.protein.faa.gz
makeblastdb -in plant.2.protein.faa -dbtype prot -parse_seqids
## Forward BLAST: Arabidopsis -> plant DB 2
blastx -query ATH_100.fasta \
-db plant.2.protein.faa \
-outfmt "6 qseqid sseqid pident evalue bitscore" \
-evalue 1e-05 \
-max_target_seqs 1 \
-num_threads 4 \
-out forward_blast.txt
Step 2: Reverse BLAST (Species 2 vs Species 1)
## Extract subject IDs from forward blast
awk '{print $2}' forward_blast.txt | sort -u > forward_hits.txt
## Extract those sequences from plant.2 database
blastdbcmd -db plant.2.protein.faa -entry_batch forward_hits.txt -out forward_hit_seqs.fasta
## Reverse BLAST: plant DB 2 hits -> Arabidopsis
makeblastdb -in Athaliana_TAIR10.cds.fa -dbtype nucl
tblastn -query forward_hit_seqs.fasta \
-db Athaliana_TAIR10.cds.fa \
-outfmt "6 qseqid sseqid pident evalue bitscore" \
-evalue 1e-05 \
-max_target_seqs 1 \
-num_threads 4 \
-out reverse_blast.txt
Step 3: Find Reciprocal Best Hits
## Create paired lists and find reciprocal matches
awk '{print $1"\t"$2}' forward_blast.txt | sort > forward_pairs.txt
awk '{print $2"\t"$1}' reverse_blast.txt | sort > reverse_pairs.txt
## Find reciprocal best hits
comm -12 forward_pairs.txt reverse_pairs.txt > reciprocal_best_hits.txt
wc -l reciprocal_best_hits.txt
Reciprocal Best BLAST Hit is a simple but effective approach for ortholog identification. For more sophisticated methods, consider tools like OrthoFinder, OrthoMCL, or InParanoid.
Practical Tips for BLAST
Memory and Performance
- Large databases: For very large databases (e.g., full nr/nt), make sure you have sufficient disk space and RAM. The nr database alone is >100 GB.
- Thread scaling: BLAST scales well up to ~8 threads. Beyond that, the performance gain diminishes.
- Query splitting: For very large query sets, split the input file and run BLAST in parallel (this is exactly what DCBLAST does).
Common Pitfalls
- Wrong database type: Using
blastnagainst a protein database (or vice versa) will produce an error. Always match your query type with the correct BLAST program. - Forgetting to build the index: Running BLAST without first running
makeblastdbwill fail. - E-value interpretation: An E-value of 0.01 means you expect 1 false positive per 100 database searches. Always consider both E-value and percent identity.
- max_target_seqs misconception:
-max_target_seqsdoes not guarantee returning the best N hits. It limits the number of hits kept during the search. For guaranteed best hits, use-max_target_seqswith a larger value and sort results afterward.
Quick Reference: Common BLAST Commands
## BLASTN: nucleotide vs nucleotide
blastn -query query.fna -db nt_db -out results.txt -outfmt 6 -evalue 1e-05 -num_threads 4
## BLASTP: protein vs protein
blastp -query query.faa -db prot_db -out results.txt -outfmt 6 -evalue 1e-05 -num_threads 4
## BLASTX: translated nucleotide vs protein
blastx -query query.fna -db prot_db -out results.txt -outfmt 6 -evalue 1e-05 -num_threads 4
## TBLASTN: protein vs translated nucleotide
tblastn -query query.faa -db nt_db -out results.txt -outfmt 6 -evalue 1e-05 -num_threads 4
## TBLASTX: translated nucleotide vs translated nucleotide
tblastx -query query.fna -db nt_db -out results.txt -outfmt 6 -evalue 1e-05 -num_threads 4
Homework: Run a BLASTX search with the full Arabidopsis CDS file against the plant protein database. Try different E-value thresholds (1e-05, 1e-10, 1e-30) and compare the number of hits. Which threshold is most appropriate and why?
DIAMOND: A Fast Alternative to BLAST
DIAMOND is an ultra-fast sequence aligner designed as a drop-in replacement for BLASTP and BLASTX. It can be 100x to 20,000x faster than NCBI BLAST while maintaining comparable sensitivity, making it ideal for large-scale searches such as metagenomic analysis or whole-genome annotation.
Why is DIAMOND Faster than BLAST?
To understand why DIAMOND is so much faster, we first need to understand the bottleneck in BLAST.
How BLAST Works (and Where It’s Slow)
BLAST processes one query at a time through the following steps:
- Break the query into short words (seeds of length W, typically 3 for protein)
- Look up each seed in the database index to find all matching positions
- Extend each seed match in both directions to produce alignments (HSPs)
- Score and filter the alignments
The bottleneck is step 2-3: for each query, BLAST must scan through the database index and perform many small extension operations. When you have thousands of queries, this process is repeated for each one independently. The disk I/O and cache misses from random database access dominate the runtime.
How DIAMOND Solves This — Four Key Strategies
DIAMOND uses a fundamentally different strategy with four major innovations:

1. Double Indexing
BLAST indexes only the database, then scans it for each query one at a time. DIAMOND indexes both the query set and the reference database simultaneously.
BLAST approach (one query at a time):
Query 1 → scan entire DB index → extend hits → output
Query 2 → scan entire DB index → extend hits → output
Query 3 → scan entire DB index → extend hits → output
... (repeated N times, many random disk accesses)
DIAMOND approach (batch all queries):
Load DB block → index ALL queries + DB block together
→ sort all seed matches → extend in sorted order → output
(sequential memory access, cache-friendly)
All seed matches between query and database are collected and sorted. Instead of randomly jumping through the database for each query, DIAMOND processes matches in a sequential, cache-friendly order. Modern CPUs are dramatically faster when accessing memory sequentially rather than randomly.
The database is loaded in large blocks into memory, and all queries are processed against each block together. This maximizes data reuse and minimizes disk I/O — the database is read from disk once per block, not once per query.
2. Spaced Seeds
Instead of using consecutive letters for seeds (like BLAST’s contiguous word), DIAMOND uses spaced seed patterns:
BLAST contiguous seed (W=3):
Query: M A S G P G G W L G
Seed: [M A S] → exact 3-letter match
DIAMOND spaced seed:
Query: M A S G P G G W L G
Seed: [# # _ # _ _ # # _ #]
M A ? G ? ? G W ? G → match at # positions, skip _ positions
Where # means “match required” and _ means “any letter allowed” (wildcard). Spaced seeds are mathematically proven to be more sensitive than contiguous seeds of the same weight. This is because:
- Contiguous seeds miss homologs when a single mutation falls inside the seed
- Spaced seeds spread required match positions across a wider region, so a single mutation is less likely to disrupt the entire seed
- Result: DIAMOND finds more true homologs with fewer seed lookups
The same spaced seed concept is used in other fast aligners like PatternHunter, BFAST, and many modern genomic tools.
3. Reduced Alphabet
DIAMOND compresses the 20 amino acid alphabet down to 11 letters by grouping biochemically similar amino acids:
Standard amino acid alphabet (20 letters):
A R N D C Q E G H I L K M F P S T W Y V
DIAMOND reduced alphabet (11 groups):
[K,R] [E,D] [Q,N] [I,L,M,V] [F,W,Y] [A] [C] [G] [H] [P] [S,T]
Why this works:
- Amino acids within each group have similar biochemical properties (charge, size, hydrophobicity)
- Substitutions within a group produce similar BLOSUM62 scores
- Seed space shrinks from 20^3 = 8,000 possible 3-letter seeds to 11^3 = 1,331 — a 6x reduction
- Fewer seeds = faster index lookups, smaller hash tables, better cache utilization
- Sensitivity is preserved because grouped amino acids would score similarly in the alignment anyway
Example: "MKLLV" in reduced alphabet
Standard: M K L L V
Reduced: [ILMV] [KR] [ILMV] [ILMV] [ILMV] → 4 1 4 4 4
A query "MRLIV" becomes:
Standard: M R L I V
Reduced: [ILMV] [KR] [ILMV] [ILMV] [ILMV] → 4 1 4 4 4 ← same seed!
The two sequences have different amino acids at positions 2, 4, and 5, but because K/R and I/L/M/V are in the same group, DIAMOND treats them as the same seed — correctly identifying them as potential homologs without needing to check every letter individually.
4. SIMD Vectorization
DIAMOND uses SIMD (Single Instruction, Multiple Data) CPU instructions to accelerate the alignment extension step. This is a hardware-level optimization that processes multiple data elements simultaneously with a single CPU instruction.
Traditional alignment (scalar, one cell at a time):
Score cell [i,j] → 1 CPU instruction → 1 result
Score cell [i,j+1] → 1 CPU instruction → 1 result
Score cell [i,j+2] → 1 CPU instruction → 1 result
Score cell [i,j+3] → 1 CPU instruction → 1 result
... 4 instructions for 4 results
SIMD alignment (vectorized, multiple cells at once):
Score cells [i,j] [i,j+1] [i,j+2] [i,j+3] → 1 CPU instruction → 4 results!
... 1 instruction for 4 results (or 8 or 16 depending on SIMD width)
Modern CPUs support several SIMD instruction sets with increasing width:
| Instruction Set | Register Width | Cells per Instruction | Speedup |
|---|---|---|---|
| SSE2 | 128-bit | 4 x 32-bit or 8 x 16-bit | ~4-8x |
| AVX2 | 256-bit | 8 x 32-bit or 16 x 16-bit | ~8-16x |
| AVX-512 | 512-bit | 16 x 32-bit or 32 x 16-bit | ~16-32x |
DIAMOND uses anti-diagonal parallelism in the Smith-Waterman dynamic programming matrix. Cells on the same anti-diagonal are independent of each other (they only depend on cells above, left, and diagonally above-left), so they can be computed simultaneously using SIMD:
Smith-Waterman DP matrix anti-diagonals:
j=0 j=1 j=2 j=3 j=4
i=0 [ 0 ] [d1] [d2 ] [d3 ] [d4 ]
i=1 [d1 ] [d2] [d3 ] [d4 ] [d5 ]
i=2 [d2 ] [d3] [d4 ] [d5 ] [d6 ]
i=3 [d3 ] [d4] [d5 ] [d6 ] [d7 ]
d1: 2 cells → computed in parallel via SIMD
d2: 3 cells → computed in parallel via SIMD
d3: 4 cells → computed in parallel via SIMD
d4: 4 cells → computed in parallel via SIMD (max parallelism)
...
Additionally, DIAMOND batches multiple alignments into a single SIMD operation. Instead of aligning one query-subject pair at a time, it packs multiple independent alignment computations into the same SIMD registers — so 8 or 16 different alignments are computed simultaneously.
You can check which SIMD instructions your CPU supports:
## Linux
grep -o 'sse2\|avx2\|avx512' /proc/cpuinfo | sort -u
## macOS
sysctl -a | grep machdep.cpu.features | grep -oi 'sse2\|avx2\|avx512'
DIAMOND automatically detects and uses the best available SIMD instruction set on your CPU. No configuration is needed — it will use AVX2 if available, falling back to SSE2 on older hardware. This is one reason DIAMOND runs faster on newer CPUs.
Summary: Four Pillars of DIAMOND Speed
| Strategy | What It Does | Speedup Factor |
|---|---|---|
| Double Indexing | Batch-processes all queries together; converts random I/O to sequential | 10-100x |
| Spaced Seeds | More sensitive seed pattern; finds more homologs per lookup | 2-3x sensitivity gain at same speed |
| Reduced Alphabet | Shrinks seed space from 8,000 to 1,331; smaller index, faster lookups | 3-6x |
| SIMD Vectorization | Computes 8-16 alignment cells simultaneously using AVX2/SSE2 | 8-16x |
The key insight is that DIAMOND trades memory for speed. By loading large chunks of data into RAM and processing all queries together, it converts random I/O into sequential I/O — and sequential access can be 100-1000x faster than random access on modern hardware. Combined with hardware-accelerated alignment via SIMD, the cumulative effect is the 100-20,000x speedup over BLAST.
DIAMOND vs BLAST Comparison
| Feature | NCBI BLAST | DIAMOND |
|---|---|---|
| Speed | Baseline | 100-20,000x faster |
| Sensitivity (default) | High | Slightly lower (~1-2% fewer hits) |
Sensitivity (--sensitive) |
High | Comparable to BLAST |
Sensitivity (--very-sensitive) |
High | Equal or better than BLAST |
| Programs supported | blastn, blastp, blastx, tblastn, tblastx | blastp, blastx |
| Nucleotide vs nucleotide | Yes (blastn) | No |
| Memory usage | Low-moderate | Higher (uses block loading) |
| Best for | Small queries, nucleotide searches | Large-scale protein searches, metagenomics |
Installation
conda activate blast
conda install -c bioconda diamond -y
Or create a separate environment:
conda create -n diamond -c bioconda -c conda-forge diamond seqkit -y
conda activate diamond
Building a DIAMOND Database
DIAMOND uses its own database format (.dmnd), which is more compact than BLAST databases:
cd ~/bch709/BLAST
## Build DIAMOND database from protein FASTA
diamond makedb --in plant.1.protein.faa --db plant.1.protein
## This creates plant.1.protein.dmnd
ls -lh plant.1.protein.dmnd
DIAMOND databases are typically 5-10x smaller than equivalent BLAST databases and faster to build.
Running DIAMOND BLASTX
The basic syntax mirrors NCBI BLAST, making it easy to switch:
cd ~/bch709/BLAST
## DIAMOND blastx (equivalent to NCBI blastx)
diamond blastx \
--query Athaliana_TAIR10.cds.fa \
--db plant.1.protein \
--out diamond_blastx_results.txt \
--outfmt 6 qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore \
--evalue 1e-05 \
--max-target-seqs 10 \
--threads 4
Running DIAMOND BLASTP
## DIAMOND blastp (equivalent to NCBI blastp)
diamond blastp \
--query your_protein.fasta \
--db plant.1.protein \
--out diamond_blastp_results.txt \
--outfmt 6 \
--evalue 1e-05 \
--max-target-seqs 5 \
--threads 4
Sensitivity Modes
DIAMOND offers multiple sensitivity levels. Higher sensitivity = slower speed but more hits found:
## Default mode (fast, good for closely related sequences)
diamond blastx --query query.fa --db db --out results_default.txt --outfmt 6
## Sensitive mode (catches more distant homologs)
diamond blastx --query query.fa --db db --out results_sensitive.txt --outfmt 6 --sensitive
## More-sensitive mode
diamond blastx --query query.fa --db db --out results_moresens.txt --outfmt 6 --more-sensitive
## Very-sensitive mode (closest to BLAST sensitivity)
diamond blastx --query query.fa --db db --out results_verysens.txt --outfmt 6 --very-sensitive
## Ultra-sensitive mode (maximum sensitivity, slowest)
diamond blastx --query query.fa --db db --out results_ultrasens.txt --outfmt 6 --ultra-sensitive
| Mode | Speed vs BLAST | Sensitivity vs BLAST |
|---|---|---|
| default | ~20,000x faster | ~98% of BLAST hits |
--sensitive |
~2,500x faster | ~99% of BLAST hits |
--more-sensitive |
~1,000x faster | ~99.5% of BLAST hits |
--very-sensitive |
~300x faster | ~99.8% of BLAST hits |
--ultra-sensitive |
~100x faster | ~100% of BLAST hits |
DIAMOND Output Formats
DIAMOND supports the same -outfmt 6 column specifiers as NCBI BLAST:
diamond blastx \
--query ATH_100.fasta \
--db plant.1.protein \
--outfmt 6 qseqid sseqid pident length evalue bitscore qcovhsp stitle \
--evalue 1e-05 \
--max-target-seqs 1 \
--threads 4 \
--out diamond_custom.txt
Additional DIAMOND-specific output formats:
| Format | Description |
|---|---|
| 0 | BLAST pairwise (human-readable) |
| 5 | BLAST XML |
| 6 | BLAST tabular (customizable columns) |
| 100 | DIAMOND alignment archive (DAA) — binary format, convertible later |
| 101 | SAM |
| 102 | Taxonomic classification |
| 103 | PAF (minimap2-compatible) |
DAA Format: DIAMOND Alignment Archive
The DAA format stores all alignment information in a compact binary file. You can convert it to other formats later without re-running the search:
## Run DIAMOND and save as DAA
diamond blastx \
--query Athaliana_TAIR10.cds.fa \
--db plant.1.protein \
--out results.daa \
--outfmt 100 \
--evalue 1e-05 \
--threads 4
## Convert DAA to tabular format
diamond view --daa results.daa --outfmt 6 --out results_tab.txt
## Convert DAA to XML format
diamond view --daa results.daa --outfmt 5 --out results.xml
## Convert with custom columns
diamond view --daa results.daa \
--outfmt 6 qseqid sseqid pident evalue stitle \
--out results_custom.txt
The DAA format is useful when you want to run a search once and then explore results in multiple formats without repeating the computation.
Key DIAMOND Options
| Option | Description | Example |
|---|---|---|
--query |
Input query FASTA file | --query query.fa |
--db |
DIAMOND database (without .dmnd) |
--db plant.1.protein |
--out |
Output file | --out results.txt |
--outfmt |
Output format and columns | --outfmt 6 qseqid sseqid pident evalue |
--evalue |
E-value threshold | --evalue 1e-05 |
--max-target-seqs |
Max hits per query | --max-target-seqs 10 |
--threads |
Number of CPU threads | --threads 4 |
--id |
Minimum percent identity | --id 80 |
--query-cover |
Minimum query coverage (%) | --query-cover 50 |
--subject-cover |
Minimum subject coverage (%) | --subject-cover 50 |
--top |
Report hits within this % of best score | --top 10 |
--block-size |
Sequence block size (billions of letters) | --block-size 2.0 |
--index-chunks |
Number of index chunks | --index-chunks 4 |
--memory-limit |
Memory limit (GB) | --memory-limit 8 |
--tmpdir |
Temp directory for large searches | --tmpdir /tmp |
Memory Tuning
For machines with limited RAM, adjust --block-size and --index-chunks:
## Low memory machine (e.g., laptop with 8 GB RAM)
diamond blastx \
--query Athaliana_TAIR10.cds.fa \
--db plant.1.protein \
--out results.txt \
--outfmt 6 \
--block-size 0.5 \
--index-chunks 4 \
--threads 4
## High memory machine (e.g., server with 64+ GB RAM)
diamond blastx \
--query Athaliana_TAIR10.cds.fa \
--db plant.1.protein \
--out results.txt \
--outfmt 6 \
--block-size 4.0 \
--index-chunks 1 \
--threads 16
--block-sizecontrols how many billions of sequence letters are processed at once. Lower values use less memory but run slower.--index-chunkssplits the database index; higher values reduce memory but increase runtime. The defaults work well for most machines with 16+ GB RAM.
Practical Example: BLAST vs DIAMOND Speed Comparison
cd ~/bch709/BLAST
## Subsample 1000 sequences for testing
seqkit sample -n 1000 Athaliana_TAIR10.cds.fa.gz > ATH_1000.fasta
## Time NCBI BLASTX
time blastx -query ATH_1000.fasta \
-db plant.1.protein.faa \
-out ncbi_blast_results.txt \
-outfmt 6 \
-evalue 1e-05 \
-max_target_seqs 5 \
-num_threads 4
## Time DIAMOND BLASTX
time diamond blastx \
--query ATH_1000.fasta \
--db plant.1.protein \
--out diamond_results.txt \
--outfmt 6 \
--evalue 1e-05 \
--max-target-seqs 5 \
--threads 4
## Compare the number of hits
echo "NCBI BLAST hits:"
wc -l ncbi_blast_results.txt
echo "DIAMOND hits:"
wc -l diamond_results.txt
When to Use DIAMOND vs BLAST
| Scenario | Recommended Tool |
|---|---|
| Small query (<100 sequences) against small DB | NCBI BLAST |
| Large query (thousands+ sequences) against protein DB | DIAMOND |
| Metagenomics / environmental sequencing | DIAMOND |
| Nucleotide vs nucleotide (blastn) | NCBI BLAST (DIAMOND doesn’t support this) |
| Need maximum sensitivity for distant homologs | NCBI BLAST or DIAMOND --ultra-sensitive |
| Genome annotation pipeline | DIAMOND |
| Quick functional annotation | DIAMOND |
| tblastn or tblastx searches | NCBI BLAST (DIAMOND doesn’t support these) |
Citation
Buchfink B, Xie C, Huson DH (2015) Fast and sensitive protein alignment using DIAMOND. Nature Methods, 12:59-60. doi:10.1038/nmeth.3176