Windows10 Local Shell
echo "export TERM=xterm-256color" >> ~/.bashrc
source ~/.bashrc
MacOS Local Shell
echo "export TERM=xterm-256color" >> ~/.bash_profile
source ~/.bash_profile
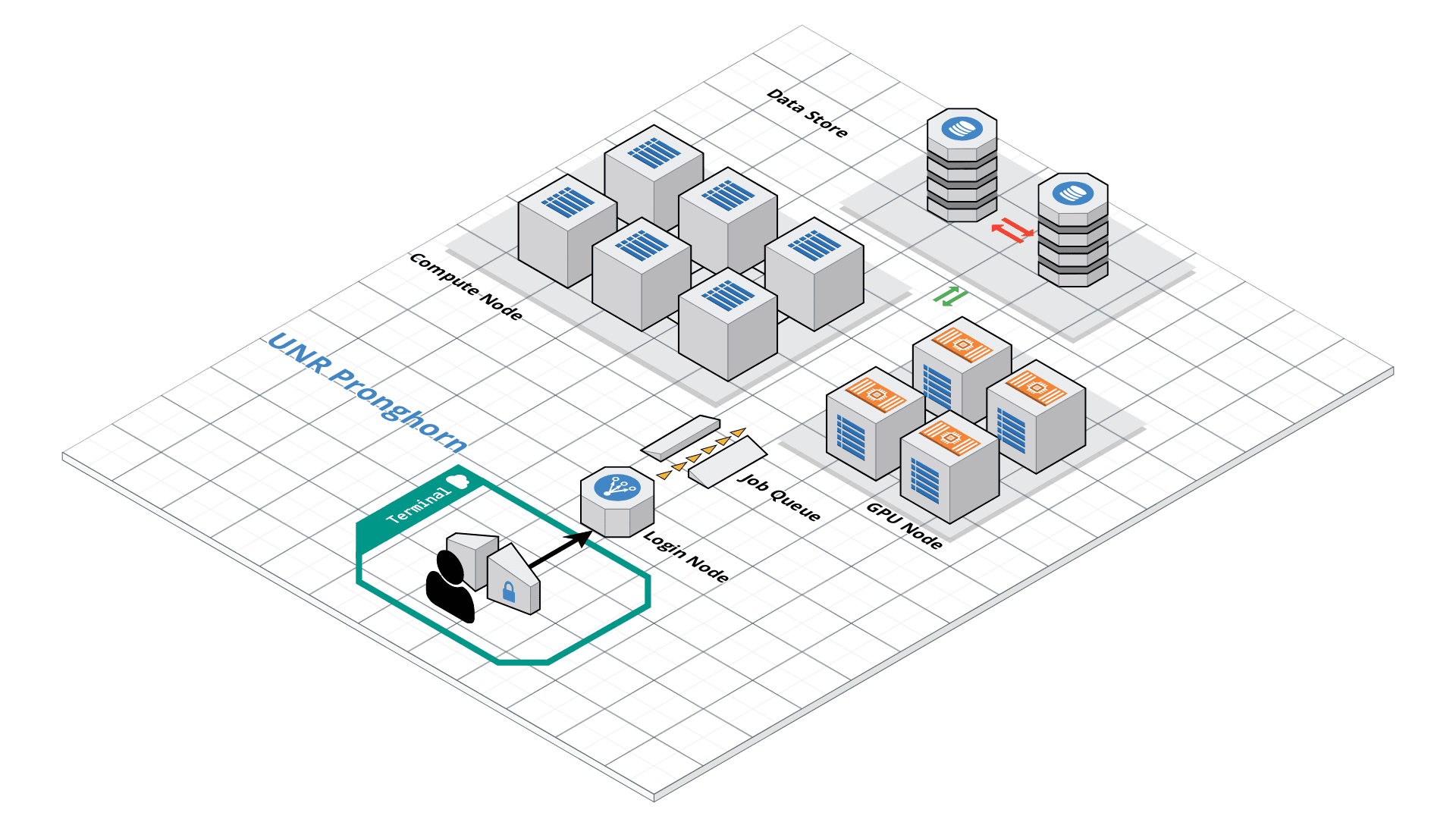
Pronghorn Storage
https://nevada.formstack.com/forms/request_research_group_access?Research_Group=BCH709
HOME WORK
- de Bruijn graph construction (10 pts)
- Draw (by hand) the de Bruijn graph for the following reads using k=3 (assume all reads are from the forward strand, no sequencing errors) AGT
ATG
CAT
GTA
GTT
TAC
TAG
TGT TTA
HOME WORK ANSWER
SSH
The ssh command is pre-installed. It means Secure Shell.
ssh <YOURID>@pronghorn.rc.unr.edu
Conda
cd ~/
rm slurm*
conda env remove --name rnaseq
wget https://www.dropbox.com/s/ed4b83m7z5ecy1y/bch709.yml
conda env create -n rnaseq -f bch709.yml
Prongrhon Shell
sed -i 's/export PS1/tty -s \&\& export PS1/g' ~/.bashrc
echo "export TERM=xterm-color" >> ~/.bashrc
source ~/.bashrc
sinfo
sinfo --all
squeue
squeue
SBATCH
Job submission
nano submit.sh
Job submission example
#!/bin/bash
#SBATCH --job-name=test
#SBATCH --cpus-per-task=16
#SBATCH --time=10:00
#SBATCH --mem=1g
#SBATCH --mail-type=begin
#SBATCH --mail-type=end
#SBATCH --mail-user=<YOUR ID>@unr.edu
echo "Hello Pronghorn"
seq 1 80000
$ chmod 775 submit.sh
$ sbatch submit.sh
sbatch: Submitted batch job <JOBID>
SCANCEL
$ scancel <JOBID>
Youtube Video for Slurm
https://www.youtube.com/watch?v=U42qlYkzP9k&list=TLrtXVJajzvonT-8qcp5ZgtKCeyN3Pe4xv
Job script generator
https://s3-us-west-2.amazonaws.com/imss-hpc/index.html
Set up Conda && activate Conda
cd ~/
conda env remove --name rnaseq
wget https://www.dropbox.com/s/ed4b83m7z5ecy1y/bch709.yml
conda env create -n rnaseq -f bch709.yml
conda activate rnaseq
Go to your storage
ls /data/gpfs/assoc/bch709/
cd /data/gpfs/assoc/bch709/
mkdir -p <YOUR_NAME>/rnaseq
Prepare your files
cp -r /data/gpfs/assoc/bch709/rawdata_rnaseq /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq
Check your files
cd /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/rawdata_rnaseq
ls fastq
Make trinity folder
cd ../
pwd
## should be /data/gpfs/assoc/bch709/<YOUR_NAME>/
mkdir Trinity
cd Trinity
###10/10/2019
From your local
export TERM=xterm-color
ssh <YOURID>@pronghorn.rc.unr.edu
Pronghorn Install directory tree
conda install -c eumetsat tree
tree
rnaseq env activation
conda activate rnaseq
conda install -c eumetsat tree
tree
Go to your storage
ls /data/gpfs/assoc/bch709/
cd /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq
Check previous fastq files
ls /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/rawdata_rnaseq/fastq

Check previous trinity files
/data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/Trinity
Running Trinity
Trinity run
pwd
## should be /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/Trinity
nano <JOBNAME>.sh
#!/bin/bash
#SBATCH --job-name=<JOBNAME>
#SBATCH --cpus-per-task=64
#SBATCH --time=2:00:00
#SBATCH --mem=100g
#SBATCH --mail-type=begin
#SBATCH --mail-type=end
#SBATCH --mail-user=<YOUR ID>@nevada.unr.edu
#SBATCH -o <JOBNAME>.out # STDOUT
#SBATCH -e <JOBNAME>.err # STDERR
Trinity --seqType fq --CPU 64 --max_memory 100G --left /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/rawdata_rnaseq/fastq/pair1.fastq.gz --right /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/rawdata_rnaseq/fastq/pair2.fastq.gz --no_normalize_reads
Submit job
sbatch <JOBNAME>.sh
Check log
cat <JOBNAME>.out
cat <JOBNAME>.err
Check the Job
tail -f <JOBNAME>.out
## or
less <JOBNAME>.out
Transcriptome and RNA-Seq Quality Evalutation
-
Examine the RNA-Seq read representation of the assembly. Ideally, at least ~80% of your input RNA-Seq reads are represented by your transcriptome assembly. The remaining unassembled reads likely corresponds to lowly expressed transcripts with insufficient coverage to enable assembly, or are low quality or aberrant reads.
-
Examine the representation of full-length reconstructed protein-coding genes, by searching the assembled transcripts against a database of known protein sequences.
-
Use BUSCO to explore completeness according to conserved ortholog content.
-
Compute the E90N50 transcript contig length - the contig N50 value based on the set of transcripts representing 90% of the expression data.
-
Compute DETONATE scores. DETONATE provides a rigorous computational assessment of the quality of a transcriptome assembly, and is useful if you want to run several assemblies using different parameter settings or using altogether different tools. That assembly with the highest DETONATE score is considered the best one.
-
Try using TransRate. TransRate generates a number of useful statistics for evaluating your transcriptome assembly. Read about TransRate here: http://genome.cshlp.org/content/26/8/1134. Note that certain statistics may be biased against the large numbers of transcripts that are very lowly expressed. Consider generating TransRate statistics for your transcriptome before and after applying a minimum expression-based filter.
-
Explore rnaQUAST a quality assessment tool for de novo transcriptome assemblies.
TransRate
Create working directory
cd /data/gpfs/assoc/bch709/<YOUR_ID>/rnaseq/
mkdir assembly_quality
cd assembly_quality
ln -s /data/gpfs/assoc/bch709/<YOUR_ID>/rnaseq/Trinity/trinity_out_dir/Trinity.fasta .
TransRate installation
conda install -c lmfaber transrate
conda install -c bioconda transrate-tools
TransRate analysis
transrate --help
nano <JOBNAME>.sh
Transrate job script
#!/bin/bash
#SBATCH --job-name=<JOBNAME>
#SBATCH --cpus-per-task=8
#SBATCH --time=2:00:00
#SBATCH --mem=16g
#SBATCH --mail-type=begin
#SBATCH --mail-type=end
#SBATCH --mail-user=<YOUR ID>@nevada.unr.edu
#SBATCH -o <JOBNAME>.out # STDOUT
#SBATCH -e <JOBNAME>.err # STDERR
transrate --assembly=Trinity.fasta --left=/data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/rawdata_rnaseq/fastq/pair1.fastq.gz --right=/data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/rawdata_rnaseq/fastq/pair2.fastq.gz
Job submission
chmod 775
sbatch <JOBNAME>.sh
Check results
cd transrate_results/
cat assemblies.csv
Open your local , please download assemblies.csv
Open this Excel
CSV = Comma-separated values
Overview of the RNA-Seq pipeline
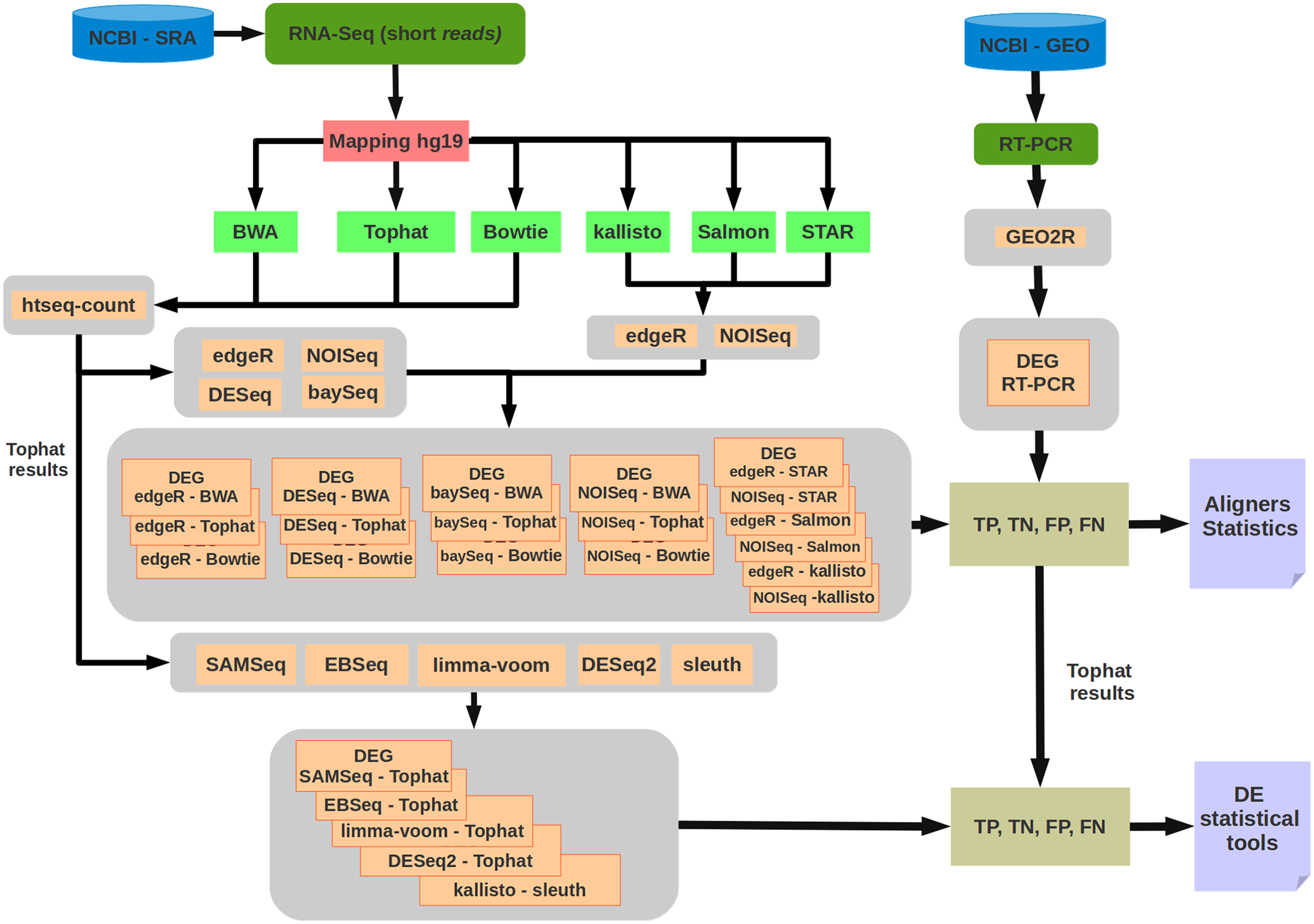
DEG
Link Assembly File
cd /data/gpfs/assoc/bch709/<YOUR_ID>/rnaseq/
mkdir DEG
cd DEG
pwd
## Your location should be /data/gpfs/assoc/bch709/<YOUR_ID>/rnaseq/DEG
ln -s /data/gpfs/assoc/bch709/<YOUR_ID>/rnaseq/Trinity/trinity_out_dir/Trinity.fasta .
Estimating Transcript Abundance
align_and_estimate_abundance.pl
The Trinity toolkit comes with a script to facilitate running your choice of the above tools to quantitate transcript abundance:
% $TRINITY_HOME/util/align_and_estimate_abundance.pl
#########################################################################
#
# --transcripts <string> transcript fasta file
# --seqType <string> fq|fa
#
# If Paired-end:
#
# --left <string>
# --right <string>
#
# or Single-end:
#
# --single <string>
# or
# --samples_file <string> tab-delimited text file indicating biological replicate relationships.
# ex.
# cond_A cond_A_rep1 A_rep1_left.fq A_rep1_right.fq
# cond_A cond_A_rep2 A_rep2_left.fq A_rep2_right.fq
# cond_B cond_B_rep1 B_rep1_left.fq B_rep1_right.fq
# cond_B cond_B_rep2 B_rep2_left.fq B_rep2_right.fq
#
# # if single-end instead of paired-end, then leave the 4th column above empty.
#
#
#
# --est_method <string> abundance estimation method.
# alignment_based: RSEM|eXpress
# alignment_free: kallisto|salmon
#
# --output_dir <string> write all files to output directory
#
#
# if alignment_based est_method:
# --aln_method <string> bowtie|bowtie2|(path to bam file) alignment method. (note: RSEM requires bowtie)
# (if you already have a bam file, you can use it here instead of rerunning bowtie)
#
# Optional:
#
# --SS_lib_type <string> strand-specific library type: paired('RF' or 'FR'), single('F' or 'R').
# (note, no strand-specific mode for kallisto)
#
# --thread_count number of threads to use (default = 4)
#
# --debug retain intermediate files
#
# --gene_trans_map <string> file containing 'gene(tab)transcript' identifiers per line.
# or
# --trinity_mode Setting --trinity_mode will automatically generate the gene_trans_map and use it.
#
#
# --prep_reference prep reference (builds target index)
#
#
########################################
#
# Parameters for single-end reads:
#
# --fragment_length <int> specify RNA-Seq fragment length (default: 200)
# --fragment_std <int> fragment length standard deviation (defalt: 80)
#
########################################
#
# bowtie-related parameters: (note, tool-specific settings are further below)
#
# --max_ins_size <int> maximum insert size (bowtie -X parameter, default: 800)
# --coordsort_bam provide coord-sorted bam in addition to the default (unsorted) bam.
#
########################################
# RSEM opts:
#
# --bowtie_RSEM <string> if using 'bowtie', default: "--all --best --strata -m 300 --chunkmbs 512"
# --bowtie2_RSEM <string> if using 'bowtie2', default: "--no-mixed --no-discordant --gbar 1000 --end-to-end -k 200 "
# --include_rsem_bam provide the RSEM enhanced bam file including posterior probabilities of read assignments.
# --rsem_add_opts <string> additional parameters to pass on to rsem-calculate-expression
#
##########################################################################
# eXpress opts:
#
# --bowtie_eXpress <string> default: "--all --best --strata -m 300 --chunkmbs 512"
# --bowtie2_eXpress <string> default: "--no-mixed --no-discordant --gbar 1000 --end-to-end -k 200 "
# --eXpress_add_opts <string> default: ""
#
##########################################################################
# kallisto opts:
#
# --kallisto_add_opts <string> default:
#
##########################################################################
#
# salmon opts:
#
# --salmon_idx_type <string> quasi|fmd (defalt: quasi)
# --salmon_add_opts <string> default:
#
#
# Example usage
#
# ## Just prepare the reference for alignment and abundance estimation
#
# /home/unix/bhaas/GITHUB/trinityrnaseq/util/align_and_estimate_abundance.pl --transcripts Trinity.fasta --est_method RSEM --aln_method bowtie --trinity_mode --prep_reference
#
# ## Run the alignment and abundance estimation (assumes reference has already been prepped, errors-out if prepped reference not located.)
#
# /home/unix/bhaas/GITHUB/trinityrnaseq/util/align_and_estimate_abundance.pl --transcripts Trinity.fasta --seqType fq --left reads_1.fq --right reads_2.fq --est_method RSEM --aln_method bowtie --trinity_mode --output_dir rsem_outdir
#
## ## prep the reference and run the alignment/estimation
#
# /home/unix/bhaas/GITHUB/trinityrnaseq/util/align_and_estimate_abundance.pl --transcripts Trinity.fasta --seqType fq --left reads_1.fq --right reads_2.fq --est_method RSEM --aln_method bowtie --trinity_mode --prep_reference --output_dir rsem_outdir
#
#########################################################################
If you have strand-specific data, be sure to include the ‘–SS_lib_type’ parameter.
Before running the above, please consider the following:
Please use the –samples_file parameter with the abundance estimation utility. This will organize your outputs so that each replicate will be organized in its own output directory named according to the corresponding replicate.
It is useful to first run ‘align_and_estimate_abundance.pl’ to only prep your reference database for alignment, using ‘–prep_reference’, and then subsequently running it on each of your sets of reads in parallel to obtain sample-specific abundance estimates.
If you quality-trimmed your reads using the –trimmomatic parameter in Trinity, you should consider using the corresponding quality-trimmed reads for the abundance estimation process outlined here. You’ll find the quality-trimmed reads in the trinity_out_dir/ with a ‘P.qtrim.gz’ extension.
Option highlight
--transcripts <string> transcript fasta file
--seqType <string> fq|fa
If Paired-end:
--left <string>
--right <string>
--aln_method bowtie|bowtie2
--est_method RSEM|kallisto|kallisto|salmon
--trinity_mode
--prep_reference
--output_dir rsem_outdir
--thread_count number of threads to use (default = 4)
RSEM
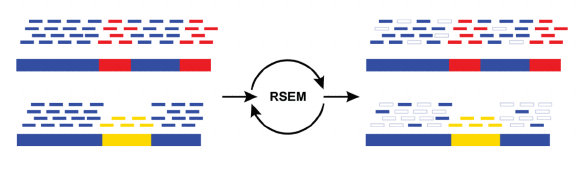 Abundance estimation via Expectation Maximization by RSEM
Abundance estimation via Expectation Maximization by RSEM
RNA-Seq reads Count Analysis
pwd
### Your current location is
## /data/gpfs/assoc/bch709/<YOUR_ID>/rnaseq/DEG
nano <JOBNAME>.sh
RNA-Seq reads Count Analysis job script
#!/bin/bash
#SBATCH --job-name=<JOBNAME>
#SBATCH --cpus-per-task=16
#SBATCH --time=2:00:00
#SBATCH --mem=16g
#SBATCH --mail-type=begin
#SBATCH --mail-type=end
#SBATCH --mail-user=<YOUR ID>@unr.edu
#SBATCH -o <JOBNAME>.out # STDOUT
#SBATCH -e <JOBNAME>.err # STDERR
align_and_estimate_abundance.pl --transcripts Trinity.fasta --seqType fq --left /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/rawdata_rnaseq/fastq/pair1.fastq.gz --right /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/rawdata_rnaseq/fastq/pair2.fastq.gz --est_method RSEM --aln_method bowtie2 --trinity_mode --prep_reference --output_dir rsem_outdir --thread_count 16
Job submission
chmod 775
sbatch <JOBNAME>.sh
Job check
squeue
Job running check
## do ```ls``` first
ls
cat <YOUR_JOB>.err
cat <YOUR_JOB>.out
RSEM results check
less rsem_outdir/RSEM.genes.results
Your current location is* /data/gpfs/assoc/bch709/wyim/rnaseq/DEG
FPKM
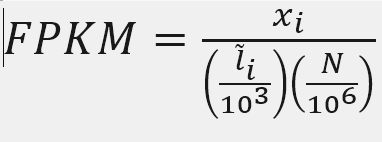
X = mapped reads count N = number of reads L = Length of transcripts
head -n 2 rsem_outdir/RSEM.genes.results
Reads count
samtools flagstat rsem_outdir/bowtie2.bam
Call Python
python
X = 861
Number_Reads_mapped = 1485483
Length = 3475
fpkm= X*(1000/Length)*(1000000/Number_Reads_mapped)
fpkm
ten to the ninth power = 10**9
fpkm=X/(Number_Reads_mapped*Length)*10**9
fpkm
FPKM
Fragments per Kilobase of transcript per million mapped reads
TPM
Transcripts Per Million
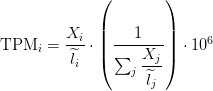
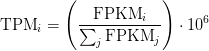
Sum of FPKM
cat rsem_outdir/RSEM.genes.results | egrep -v FPKM | awk '{ sum+=$7} END {print sum}'
TPM calculation from FPKM
FPKM = 180.61
SUM_FPKM = 646089
TPM=(FPKM/SUM_FPKM)*10**6
TPM
TPM calculation from reads count
cat rsem_outdir/RSEM.genes.results | egrep -v FPKM | awk '{ sum+=$5/$3} END {print sum}'
sum_count_per_length = 714.037
TPM = (X/Length)*(1/sum_count_per_length )*10**6
Paper read
Li et al., 2010, RSEM Dillies et al., 2013
HOME WORK
File download
$ mkdir -p /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/homework
$ cd /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/homework
Download below files
mkdir fastq
cd fastq
pwd
#current directory should be /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/homework/fastq
- use
wgethttps://www.dropbox.com/s/mzvempzve7uuwoa/KRWTD1_1.fastq.gz https://www.dropbox.com/s/uv2a10w9wj9tcww/KRWTD1_2.fastq.gz https://www.dropbox.com/s/ra0s10g2nag6axp/KRWTD2_1.fastq.gz https://www.dropbox.com/s/jao5tb8a1hnzqw4/KRWTD2_2.fastq.gz https://www.dropbox.com/s/4gwhz0t1d32cdnw/KRWTD3_1.fastq.gz https://www.dropbox.com/s/wp2uk0nafdb74wp/KRWTD3_2.fastq.gz https://www.dropbox.com/s/ctzo9k9n8qdpvio/KRWTW1_1.fastq.gz https://www.dropbox.com/s/psiak4r2910sjsc/KRWTW1_2.fastq.gz https://www.dropbox.com/s/so4zeuyqz64m80z/KRWTW2_1.fastq.gz https://www.dropbox.com/s/2ggf2xdiydtehdw/KRWTW2_2.fastq.gz https://www.dropbox.com/s/7bfgcq69cymb5yj/KRWTW3_1.fastq.gz https://www.dropbox.com/s/lfif4qnbhnfes26/KRWTW3_2.fastq.gz
Run trimming
$ cd /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/homework/fastq
$ nano <JOB_NAME>.sh
Submit trimming job with following criteria
- SBATCH
--time=2:00:00 --cpus-per-task=16 --mem-per-cpu=10g - trim_galore
trim_galore --paired --three_prime_clip_R1 20 --three_prime_clip_R2 20 --cores 16 --max_n 40 --gzip -o /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/homework/trim
Mock SBATCH example is below
#!/bin/bash
#SBATCH --job-name=<JOB_NAME>
#SBATCH --cpus-per-task=1
#SBATCH --time=2:00:00
#SBATCH --mem-per-cpu=1g
#SBATCH --mail-type=begin
#SBATCH --mail-type=end
#SBATCH --mail-user=<YOUR ID>@unr.edu
trim_galore --paired --three_prime_clip_R1 20 --three_prime_clip_R2 20 --cores 16 --max_n 40 --gzip -o /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/homework/trim /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/homework/fastq/KRWTD1_1.fastq.gz /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/homework/fastq/KRWTD1_2.fastq.gz
Job submission Requirement
chmod 775 <JOB_NAME>.sh
sbatch <JOB_NAME>.sh
Need to do all of samples
KRWTD1_1.fastq.gz KRWTD1_2.fastq.gz
KRWTD2_1.fastq.gz KRWTD2_2.fastq.gz
KRWTD3_1.fastq.gz KRWTD3_2.fastq.gz
KRWTW1_1.fastq.gz KRWTW1_2.fastq.gz
KRWTW2_1.fastq.gz KRWTW2_2.fastq.gz
KRWTW3_1.fastq.gz KRWTW3_2.fastq.gz
Run FastQC on Pronghorn by using SBATCH
- SBATCH
--time=2:00:00 --cpus-per-task=16 --mem-per-cpu=10g - fastqc
You can do everything at once.
fastqc /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/homework/trim/*.gz fastqc /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/homework/fastq/*.gz
PLEASE CHECK YOUR MULTIQC
Merge gz file (Pair1 and Pair2)
cd /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/homework/trim
### For example
zcat /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/homework/trim/*.1.fq.gz >> merged_R1.fastq
zcat /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/homework/trim/*.2.fq.gz >> merged_R2.fastq
Trinity run
mkdir /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/homework/Trinity
cd /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/homework/Trinity
Submit Trinity job with following criteria
#!/bin/bash
#SBATCH --job-name=<JOB_NAME>
#SBATCH --cpus-per-task=64
#SBATCH --time=2:00:00
#SBATCH --mem=100g
#SBATCH --mail-type=begin
#SBATCH --mail-type=end
#SBATCH --mail-user=<YOUR ID>@unr.edu
#SBATCH -o <JOB_NAME>.out # STDOUT
#SBATCH -e <JOB_NAME>.err # STDERR
Trinity --seqType fq --CPU 64 --max_memory 100G --left <PAIR1_MERGED_FILE_LOCATION> --right <PAIR2_MERGED_FILE_LOCATION>
MultiQC
cd /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/homework/
multiqc . -n <YOUR_NAME> --exclude rsem --exclude gatk
Please send me the results
cd /data/gpfs/assoc/bch709/<YOUR_NAME>/rnaseq/homework/Trinity/
egrep -c ">" trinity_out_dir/Trinity.fasta >> <YOUR_NAME>.trinity.stat
TrinityStats.pl trinity_out_dir/Trinity.fasta >> <YOUR_NAME>.trinity.stat
Use SCP for downloading and send me the file
***
** When you have an ERROR, please send me your history. “histoty » myhistory.txt” **
Feedback required
https://forms.gle/DZ6vLkJ4iJpryK5Z6