Environment creation
conda create -n BCH709_RNASeq -c bioconda -c conda-forge -c anaconda python=3.7 mamba
conda activate BCH709_RNASeq
mamba install -c bioconda -c conda-forge -c anaconda trim-galore=0.6.7 sra-tools=2.11.0 STAR htseq=1.99.2 subread=2.0.1 multiqc=1.11 snakemake=7.5.0 parallel-fastq-dump=0.6.7 bioconductor-tximport samtools=1.14 r-ggplot2 trinity=2.13.2 hisat2 bioconductor-qvalue sambamba graphviz gffread tpmcalculator lxml rsem
Slurm

Slurm provides resource management for the processors allocated to a job, so that multiple job steps can be simultaneously submitted and queued until there are available resources within the job’s allocation.
Mouse RNA-Seq
https://www.sciencedirect.com/science/article/pii/S2211124722011111
Working directory (Pronghorn)
echo $USER
cd /data/gpfs/assoc/bch709-3/${USER}
mkdir mouse
mkdir /data/gpfs/assoc/bch709-3/${USER}/mouse/fastq
mkdir /data/gpfs/assoc/bch709-3/${USER}/mouse/ref
mkdir /data/gpfs/assoc/bch709-3/${USER}/mouse/trim
mkdir /data/gpfs/assoc/bch709-3/${USER}/mouse/bam
mkdir /data/gpfs/assoc/bch709-3/${USER}/mouse/readcount
mkdir /data/gpfs/assoc/bch709-3/${USER}/mouse/DEG
Reference Download
https://hgdownload.soe.ucsc.edu/downloads.html
### change working directory
cd /data/gpfs/assoc/bch709-3/${USER}/mouse/ref
### download
wget https://hgdownload.soe.ucsc.edu/goldenPath/mm39/bigZips/mm39.fa.gz
wget https://hgdownload.soe.ucsc.edu/goldenPath/mm39/bigZips/genes/refGene.gtf.gz
### decompress
gunzip mm39.fa.gz
gunzip refGene.gtf.gz
### Copy templet
cp /data/gpfs/assoc/bch709-3/Course_materials/mouse/run.sh /data/gpfs/assoc/bch709-3/${USER}/mouse/ref/ref_build.sh
STAR aligner reference build example
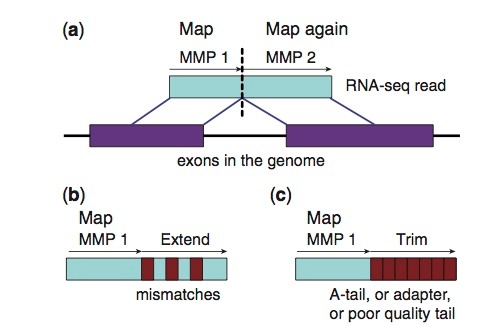
STAR --runThreadN [CPU] --runMode genomeGenerate --genomeDir . --genomeFastaFiles [GENOMEFASTA] --sjdbGTFfile [GENOME_GTF] --sjdbOverhang 99 --genomeSAindexNbases 12
Bioinformatics. 2013 Jan; 29(1): 15–21 10.1093/bioinformatics/bts635
STAR aligner reference build on Pronghorn
#open text editor
nano ref_build.sh
# Add below command to ref_build.sh
STAR --runThreadN 4 --runMode genomeGenerate --genomeDir . --genomeFastaFiles mm39.fa --sjdbGTFfile refGene.gtf --sjdbOverhang 99 --genomeSAindexNbases 12
Submit job to HPC
#submit job
sbach ref_build.sh
#check job
squeue
FASTQ file
cd /data/gpfs/assoc/bch709-3/${USER}/mouse/
### Link file (without copy)
ln -s /data/gpfs/assoc/bch709-3/Course_materials/mouse/fastq/* /data/gpfs/assoc/bch709-3/${USER}/mouse/fastq
ls /data/gpfs/assoc/bch709-3/${USER}/mouse/fastq
Create file list
cd /data/gpfs/assoc/bch709-3/${USER}/mouse/fastq
ls -1 *.gz
ls -1 *.gz | sed 's/_R.\.fastq\.gz//g'
ls -1 *.gz | sed 's/_R.\.fastq\.gz//g' | sort -u
ls -1 *.gz | sed 's/_R.\.fastq\.gz//g' | sort -u >> /data/gpfs/assoc/bch709-3/${USER}/mouse/filelist
cat /data/gpfs/assoc/bch709-3/${USER}/mouse/filelist
Regular expression
https://regex101.com/
Trim reads
trim_galore --paired --three_prime_clip_R1 [integer] --three_prime_clip_R2 [integer] --cores [integer] --max_n [integer] --fastqc --gzip -o /data/gpfs/assoc/bch709-3/${USER}/mouse/trim {READ_R1} {READ_R2}
Trim reads example
trim_galore --paired --three_prime_clip_R1 5 --three_prime_clip_R2 5 --cores 2 --max_n 40 --fastqc --gzip -o /data/gpfs/assoc/bch709-3/${USER}/mouse/trim {READ_R1} {READ_R2}
Prepare templet
cp /data/gpfs/assoc/bch709-3/Course_materials/mouse/run.sh /data/gpfs/assoc/bch709-3/${USER}/mouse/fastq/trim.sh
sed -i "s/\-\-cpus\-per\-task\=2/\-\-cpus\-per\-task\=4/g; s/\[NAME\]/Trim/g; s/\[youremail\]/${USER}\@unr.edu\,${USER}\@nevada.unr.edu/g" /data/gpfs/assoc/bch709-3/${USER}/mouse/fastq/trim.sh
Edit templet
nano /data/gpfs/assoc/bch709-3/${USER}/mouse/fastq/trim.sh
Batch submission
# Check file list
cat ../filelist
# Loop file list
### Add Forward read to variable
### Add reverse read from forward read name substitution
for i in `cat ../filelist`
do
read1=${i}_R1.fastq.gz
read2=${read1//_R1.fastq.gz/_R2.fastq.gz}
echo $read1 $read2
done
# Loop file list
### add file name from variable to trim-galore
for i in `cat ../filelist`
do
read1=${i}_R1.fastq.gz
read2=${read1//_R1.fastq.gz/_R2.fastq.gz}
echo $read1 $read2
echo "trim_galore --paired --three_prime_clip_R1 5 --three_prime_clip_R2 5 --cores 2 --max_n 40 --fastqc --gzip -o /data/gpfs/assoc/bch709-3/${USER}/mouse/trim $read1 $read2"
done
### merge trim-galore command and trim.sh
for i in `cat ../filelist`
do
read1=${i}_R1.fastq.gz
read2=${read1//_R1.fastq.gz/_R2.fastq.gz}
echo $read1 $read2
echo "trim_galore --paired --three_prime_clip_R1 5 --three_prime_clip_R2 5 --cores 2 --max_n 40 --fastqc --gzip -o /data/gpfs/assoc/bch709-3/${USER}/mouse/trim $read1 $read2" | cat trim.sh -
done
### THIS IS FINAL one
### add trim-galore command and trim.sh to new file
for i in `cat ../filelist`
do
read1=${i}_R1.fastq.gz
read2=${read1//_R1.fastq.gz/_R2.fastq.gz}
echo $read1 $read2
echo "trim_galore --paired --three_prime_clip_R1 5 --three_prime_clip_R2 5 --cores 2 --max_n 40 --fastqc --gzip -o /data/gpfs/assoc/bch709-3/${USER}/mouse/trim $read1 $read2" | cat trim.sh - > ${i}_trim.sh
done
Batch submission
ls *.sh
ls -1 *.sh
### Loop *.sh printing
for i in `ls -1 *.sh`
do
echo $i
done
### Loop *.sh submission
for i in `ls -1 *.sh`
do
sbatch $i
done
Check submission
squeue -u ${USER}
RNA-Seq Alignment
#### Move to trim folder
cd /data/gpfs/assoc/bch709-3/${USER}/mouse/trim
#### Copy templet
cp /data/gpfs/assoc/bch709-3/Course_materials/mouse/run.sh /data/gpfs/assoc/bch709-3/${USER}/mouse/trim/mapping.sh
sed -i "s/16g/64g/g; s/\-\-cpus\-per\-task\=2/\-\-cpus\-per\-task\=4/g; s/\[NAME\]/Trim/g; s/\[youremail\]/${USER}\@unr.edu\,${USER}\@nevada.unr.edu/g" /data/gpfs/assoc/bch709-3/${USER}/mouse/trim/mapping.sh
#### Edit templet
nano mapping.sh
Check output
ls -algh /data/gpfs/assoc/bch709-3/${USER}/mouse/trim
Output example
[FILENAME]_R1_val_1.fq.gz [FILENAME]_R2_val_2.fq.gz
STAR RNA-Seq read alignment example
STAR --runMode alignReads --runThreadN [CPU_NUMBER] --outFilterMultimapNmax 100 --alignIntronMin 25 --alignIntronMax 50000 --quantMode TranscriptomeSAM GeneCounts --genomeDir [GENOME_DIR] --readFilesCommand gunzip -c --readFilesIn [FORWARD_READ] [REVERSE_READ] --outSAMtype BAM SortedByCoordinate --outFileNamePrefix [BAMFILENAME_LOCATION]
STAR RNA-Seq alignment
STAR --runMode alignReads --runThreadN 4 --outFilterMultimapNmax 100 --alignIntronMin 25 --alignIntronMax 50000 --quantMode TranscriptomeSAM GeneCounts --genomeDir /data/gpfs/assoc/bch709-3/${USER}/mouse/ref --readFilesCommand gunzip -c --readFilesIn /data/gpfs/assoc/bch709-3/${USER}/mouse/trim/[FILENAME]_R1_val_1.fq.gz /data/gpfs/assoc/bch709-3/${USER}/mouse/trim/[FILENAME]_R2_val_2.fq.gz --outSAMtype BAM SortedByCoordinate --outFileNamePrefix /data/gpfs/assoc/bch709-3/${USER}/mouse/bam/[FILENAME].bam
STAR RNA-Seq alignment batch file test
for i in `cat ../filelist`
do
read1=${i}_R1_val_1.fq.gz
read2=${read1//_R1_val_1.fq.gz/_R2_val_2.fq.gz}
echo $read1 $read2
echo "STAR --runMode alignReads --runThreadN 4 --outFilterMultimapNmax 100 --alignIntronMin 25 --alignIntronMax 50000 --genomeDir /data/gpfs/assoc/bch709-3/${USER}/mouse/ref --readFilesCommand gunzip -c --readFilesIn /data/gpfs/assoc/bch709-3/${USER}/mouse/trim/${read1} /data/gpfs/assoc/bch709-3/${USER}/mouse/trim/${read2} --outSAMtype BAM SortedByCoordinate --outFileNamePrefix /data/gpfs/assoc/bch709-3/${USER}/mouse/bam/${i}.bam"
done
STAR RNA-Seq alignment batch file
for i in `cat ../filelist`
do
read1=${i}_R1_val_1.fq.gz
read2=${read1//_R1_val_1.fq.gz/_R2_val_2.fq.gz}
echo $read1 $read2
echo "STAR --runMode alignReads --runThreadN 4 --outFilterMultimapNmax 100 --alignIntronMin 25 --alignIntronMax 50000 --genomeDir /data/gpfs/assoc/bch709-3/${USER}/mouse/ref --readFilesCommand gunzip -c --readFilesIn /data/gpfs/assoc/bch709-3/${USER}/mouse/trim/${read1} /data/gpfs/assoc/bch709-3/${USER}/mouse/trim/${read2} --outSAMtype BAM SortedByCoordinate --outFileNamePrefix /data/gpfs/assoc/bch709-3/${USER}/mouse/bam/${i}.bam" | cat mapping.sh - > ${i}_mapping.sh
done
Job submission dependency
squeue --noheader --format %i --user ${USER}
squeue --noheader --format %i --user ${USER} | tr '\n' ':'
Job submission dependency on Mapping
jobid=$(squeue --noheader --format %i --user ${USER} | tr '\n' ':')1
for i in `ls -1 *_mapping.sh`
do
sbatch --dependency=afterany:${jobid} $i
done
featureCounts -o [output] -T [threads] -Q 1 -p -M -g gene_id -a [GTF] [BAMs]
FeatureCounts execute location
#### Move to trim folder
cd /data/gpfs/assoc/bch709-3/${USER}/mouse/bam
ls -1 *.bam
#### Copy templet
cp /data/gpfs/assoc/bch709-3/Course_materials/mouse/run.sh /data/gpfs/assoc/bch709-3/${USER}/mouse/bam/count.sh
sed -i "s/16g/64g/g; s/\-\-cpus\-per\-task\=2/\-\-cpus\-per\-task\=4/g; s/\[NAME\]/Count/g; s/\[youremail\]/${USER}\@unr.edu\,${USER}\@nevada.unr.edu/g" /data/gpfs/assoc/bch709-3/${USER}/mouse/bam/count.sh
FeatureCounts read bam file
cd /data/gpfs/assoc/bch709-3/${USER}/mouse/bam
ls -1 *.sortedByCoord.out.bam
ls -1 *.sortedByCoord.out.bam| tr '\n' ' '
Edit templet
nano count.sh
#paste this to count.sh
featureCounts -o /data/gpfs/assoc/bch709-3/${USER}//mouse/readcount/featucount -T 4 -Q 1 -p -M -g gene_id -a /data/gpfs/assoc/bch709-3/${USER}/mouse/ref/refGene.gtf $(for i in `cat /data/gpfs/assoc/bch709-3/wyim/mouse/filelist`; do echo ${i}.bamAligned.sortedByCoord.out.bam| tr '\n' ' ';done)
Job submission dependency
squeue --noheader --format %i --user ${USER}
Submit
jobid=$(squeue --noheader --format %i --user ${USER} | tr '\n' ':')1
sbatch --dependency=afterany:${jobid} count.sh
Human RNA-Seq
Transcriptome alterations in myotonic dystrophy frontal cortex
Environment activation
conda activate BCH709_RNASeq
Working directory (Pronghorn)
echo $USER
cd /data/gpfs/assoc/bch709-3/${USER}
mkdir human
mkdir /data/gpfs/assoc/bch709-3/${USER}/human/fastq
mkdir /data/gpfs/assoc/bch709-3/${USER}/human/ref
mkdir /data/gpfs/assoc/bch709-3/${USER}/human/trim
mkdir /data/gpfs/assoc/bch709-3/${USER}/human/bam
mkdir /data/gpfs/assoc/bch709-3/${USER}/human/readcount
mkdir /data/gpfs/assoc/bch709-3/${USER}/human/DEG
Reference Download
https://www.ncbi.nlm.nih.gov/genome/guide/human/
### change working directory
cd /data/gpfs/assoc/bch709-3/${USER}/human/ref
### download
wget https://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/genes/hg38.refGene.gtf.gz
wget https://ftp.ncbi.nlm.nih.gov/refseq/H_sapiens/annotation/GRCh38_latest/refseq_identifiers/GRCh38_latest_genomic.fna.gz
### decompress
gunzip GRCh38_latest_genomic.fna.gz
gunzip hg38.refGene.gtf.gz
STAR reference build
STAR aligner reference build on Pronghorn
### Copy templet
cp /data/gpfs/assoc/bch709-3/Course_materials/human/run.sh /data/gpfs/assoc/bch709-3/${USER}/human/ref/ref_build.sh
#open text editor
### PLEASE RENAME EMAIL AND JOB NAME
sed -i "s/16g/64g/g; s/\-\-cpus\-per\-task\=2/\-\-cpus\-per\-task\=4/g; s/\[NAME\]/ref_build/g; s/\[youremail\]/${USER}\@unr.edu\,${USER}\@nevada.unr.edu/g" /data/gpfs/assoc/bch709-3/${USER}/human/ref/ref_build.sh
nano ref_build.sh
# Add below command to ref_build.sh
STAR --runThreadN 4 --runMode genomeGenerate --genomeDir . --genomeFastaFiles GRCh38_latest_genomic.fna --sjdbGTFfile GRCh38_latest_genomic.gtf --sjdbOverhang 99 --genomeSAindexNbases 12
Submit job to HPC
#submit job
sbach ref_build.sh
#check job
squeue -u ${USER}
FASTQ file
cd /data/gpfs/assoc/bch709-3/${USER}/human/
### Link file (without copy)
ln -s /data/gpfs/assoc/bch709-3/Course_materials/human/fastq/* /data/gpfs/assoc/bch709-3/${USER}/human/fastq
ls /data/gpfs/assoc/bch709-3/${USER}/human/fastq
Create file list
cd /data/gpfs/assoc/bch709-3/${USER}/human/fastq
ls -1 *.gz
ls -1 *.gz | sed 's/_R.\.fastq\.gz//g'
ls -1 *.gz | sed 's/_R.\.fastq\.gz//g' | sort -u
ls -1 *.gz | sed 's/_R.\.fastq\.gz//g' | sort -u > /data/gpfs/assoc/bch709-3/${USER}/human/filelist
cat /data/gpfs/assoc/bch709-3/${USER}/human/filelist
Regular expression
https://regex101.com/
Trim reads
Prepare templet
cp /data/gpfs/assoc/bch709-3/Course_materials/human/run.sh /data/gpfs/assoc/bch709-3/${USER}/human/fastq/trim.sh
sed -i "s/16g/64g/g; s/\-\-cpus\-per\-task\=2/\-\-cpus\-per\-task\=4/g; s/\[NAME\]/Trim/g; s/\[youremail\]/${USER}\@unr.edu\,${USER}\@nevada.unr.edu/g" /data/gpfs/assoc/bch709-3/${USER}/human/fastq/trim.sh
Edit templet
nano /data/gpfs/assoc/bch709-3/${USER}/human/fastq/trim.sh
Batch submission
# Check file list
cat ../filelist
nano trim.sh
# Loop file list
### Add Forward read to variable
### Add reverse read from forward read name substitution
### add file name from variable to trim-galore
### merge trim-galore command and trim.sh
### add trim-galore command and trim.sh to new file
for i in `cat ../filelist`
do
read1=${i}_R1.fastq.gz
read2=${read1//_R1.fastq.gz/_R2.fastq.gz}
echo "trim_galore --paired --three_prime_clip_R1 5 --three_prime_clip_R2 5 --cores 2 --max_n 40 --fastqc --gzip -o /data/gpfs/assoc/bch709-3/${USER}/human/trim $read1 $read2" | cat trim.sh - > ${i}_trim.sh
echo "$read1 $read2 trim file has been created."
done
Batch submission
ls -1 *_trim.sh
### Loop *.sh submission
for i in `ls -1 *_trim.sh`
do
sbatch $i
done
Check submission
squeue -u ${USER}
RNA-Seq Alignment
#### Move to trim folder
cd /data/gpfs/assoc/bch709-3/${USER}/human/trim
#### Copy templet
cp /data/gpfs/assoc/bch709-3/Course_materials/human/run.sh /data/gpfs/assoc/bch709-3/${USER}/human/trim/mapping.sh
sed -i "s/16g/64g/g; s/\-\-cpus\-per\-task\=2/\-\-cpus\-per\-task\=4/g; s/\[NAME\]/Mapping/g; s/\[youremail\]/${USER}\@unr.edu\,${USER}\@nevada.unr.edu/g" /data/gpfs/assoc/bch709-3/${USER}/human/trim/mapping.sh
#### Edit templet
nano mapping.sh
Check output
ls -algh /data/gpfs/assoc/bch709-3/${USER}/human/trim
Output example
[FILENAME]_R1_val_1.fq.gz [FILENAME]_R2_val_2.fq.gz
STAR RNA-Seq alignment
STAR --runMode alignReads --runThreadN 4 --outFilterMultimapNmax 100 --alignIntronMin 25 --alignIntronMax 50000 --quantMode TranscriptomeSAM GeneCounts --genomeDir /data/gpfs/assoc/bch709-3/${USER}/human/ref --readFilesCommand gunzip -c --readFilesIn /data/gpfs/assoc/bch709-3/${USER}/human/trim/[FILENAME]_R1_val_1.fq.gz /data/gpfs/assoc/bch709-3/${USER}/human/trim/[FILENAME]_R2_val_2.fq.gz --outSAMtype BAM SortedByCoordinate --outFileNamePrefix /data/gpfs/assoc/bch709-3/${USER}/human/bam/[FILENAME].bam
STAR RNA-Seq alignment batch file
cd /data/gpfs/assoc/bch709-3/${USER}/human/trim
for i in `cat ../filelist`
do
read1=${i}_R1_val_1.fq.gz
read2=${read1//_R1_val_1.fq.gz/_R2_val_2.fq.gz}
echo $read1 $read2
echo "STAR --runMode alignReads --runThreadN 4 --outFilterMultimapNmax 100 --alignIntronMin 25 --alignIntronMax 50000 --genomeDir /data/gpfs/assoc/bch709-3/${USER}/human/ref --outSAMtype BAM SortedByCoordinate --readFilesCommand gunzip -c --readFilesIn /data/gpfs/assoc/bch709-3/${USER}/human/trim/${read1} /data/gpfs/assoc/bch709-3/${USER}/human/trim/${read2} --outFileNamePrefix /data/gpfs/assoc/bch709-3/${USER}/human/bam/${i}.bam" | cat mapping.sh - > ${i}_mapping.sh
done
Job submission dependency
squeue --noheader --format %i --user ${USER}
squeue --noheader --format %i --user ${USER} | tr '\n' ':'
Job submission dependency on Trim
jobid=$(squeue --noheader --format %i --user ${USER} | tr '\n' ':')1
for i in `ls -1 *_mapping.sh`
do
sbatch --dependency=afterany:${jobid} $i
done
FeatureCounts
Bioinformatics, Volume 30, Issue 7, 1 April 2014, Pages 923–930

featureCounts -o [output] -T [threads] -Q 1 -p -M -g gene_id -a [GTF] [BAMs]
FeatureCounts location
#### Move to trim folder
cd /data/gpfs/assoc/bch709-3/${USER}/human/bam
#### Copy templet
cp /data/gpfs/assoc/bch709-3/Course_materials/human/run.sh /data/gpfs/assoc/bch709-3/${USER}/human/bam/count.sh
sed -i "s/16g/64g/g; s/\-\-cpus\-per\-task\=2/\-\-cpus\-per\-task\=4/g; s/\[NAME\]/Count/g; s/\[youremail\]/${USER}\@unr.edu\,${USER}\@nevada.unr.edu/g" /data/gpfs/assoc/bch709-3/${USER}/human/bam/count.sh
FeatureCounts command to count.sh
LOOP example
cd /data/gpfs/assoc/bch709-3/${USER}/human/bam
ls -1 *.bam
for i in `cat /data/gpfs/assoc/bch709-3/wyim/human/filelist`
do
echo ${i}.bamAligned.sortedByCoord.out.bam | tr '\n' ' '
done
FeatureCount
echo "featureCounts -o /data/gpfs/assoc/bch709-3/${USER}//mouse/readcount/featucount -T 4 -Q 1 -p -M -g gene_id -a /data/gpfs/assoc/bch709-3/${USER}/human/ref/GRCh38_latest_genomic.gtf $(for i in `cat /data/gpfs/assoc/bch709-3/wyim/human/filelist`; do echo ${i}.bamAligned.sortedByCoord.out.bam| tr '\n' ' ';done)" >> count.sh
Job submission dependency
squeue --noheader --format %i --user ${USER}
squeue --noheader --format %i --user ${USER} | tr '\n' ':'
Job submission dependency on Align
cd /data/gpfs/assoc/bch709-3/${USER}/human/bam
jobid=$(squeue --noheader --format %i --user ${USER} | tr '\n' ':')1
sbatch --dependency=afterany:${jobid} count.sh