Course Evaluation
Students will have access to course evaluation You can log in with your NetID to http://www.unr.edu/evaluate and check live updating response rates for your course evaluations. Our institutional goal is to achieve an 85% response rate for all evaluations, and to help us achieve that, we rely on you as well as the students. Students will have access to them until 11:59 PM on Wed, May 6, 2020 PDT.
If we can achieve 100% response rate for evaluation, I will give you additional points for all of you.
RNA reads alignment
The alignment process consists of choosing an appropriate reference genome to map our reads against and performing the read alignment using one of several splice-aware alignment tools such as STAR or HISAT2. The choice of aligner is often a personal preference and also dependent on the computational resources that are available to you.
Location
/data/gpfs/assoc/bch709/<YOURID>/genomernaseq
Environment
conda create -n genomernaseq
conda activate genomernaseq
conda install -c bioconda bioconductor-qvalue bioconductor-edger bioconductor-deseq2 starseqr samtools gffread star subread pandas -y
conda install -c r -c bioconda r-fastcluster -y
conda install -c bioconda -c conda-forge openblas openssl=1.0 -y
Reads preparation
/data/gpfs/assoc/bch709/Course_material/2020/RNASeq_trimmed_fastq
Genome annotation preparation
/data/gpfs/assoc/bch709/Course_material/2020/genome/bch709_assembly.fasta
/data/gpfs/assoc/bch709/Course_material/2020/genome/bch709.gff
GTF format
The Gene Transfer Format (GTF) is a widely used format for storing gene annotations. You can obtain GTF files easily from the UCSC table browser and Ensembl
Fields must be tab-separated. Also, all but the final field in each feature line must contain a value; “empty” columns should be denoted with a ‘.’
- seqname - name of the chromosome or scaffold; chromosome names can be given with or without the ‘chr’ prefix. Important note: the seqname must be one used within Ensembl, i.e. a standard chromosome name or an Ensembl identifier such as a scaffold ID, without any additional content such as species or assembly. See the example GFF output below.
- source - name of the program that generated this feature, or the data source (database or project name)
- feature - feature type name, e.g. Gene, Variation, Similarity
- start - Start position of the feature, with sequence numbering starting at 1.
- end - End position of the feature, with sequence numbering starting at 1.
- score - A floating point value.
- strand - defined as + (forward) or - (reverse).
- frame - One of ‘0’, ‘1’ or ‘2’. ‘0’ indicates that the first base of the feature is the first base of a codon, ‘1’ that the second base is the first base of a codon, and so on..
- attribute - A semicolon-separated list of tag-value pairs, providing additional information about each feature.
GFF3 to GTF
gffread --help
gffread bch709.gff -T -o bch709.gtf
STAR Aligner
We will align our reads to the reference genome using STAR (Spliced Transcripts Alignment to a Reference). STAR is an aligner designed to specifically address many of the challenges of RNA-seq data mapping using a strategy to account for spliced alignments.
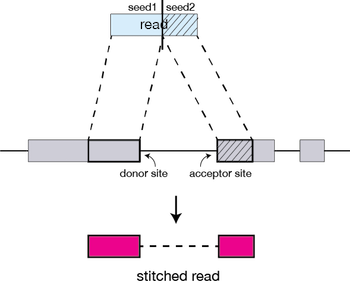
For every read that STAR aligns, STAR will search for the longest sequence that exactly matches one or more locations on the reference genome. These longest matching sequences are called the Maximal Mappable Prefixes (MMPs)
Genome sequence preparation
Now that you have the genome and annotation files, you will create a genome index using the following script:
#!/bin/bash
#SBATCH --job-name=star
#SBATCH --cpus-per-task=2
#SBATCH --time=12:00:00
#SBATCH --mem=8g
#SBATCH --mail-type=all
#SBATCH --mail-user=<EMAIL>@unr.edu
#SBATCH -o star.out # STDOUT
#SBATCH -e star.err # STDERR
#SBATCH -p cpu-s2-core-0
#SBATCH -A cpu-s2-bch709-0
mkdir reference
STAR --runMode genomeGenerate --genomeDir /data/gpfs/assoc/bch709/<YOURID>/genomernaseq/reference --genomeFastaFiles /data/gpfs/assoc/bch709/<YOURID>/genomernaseq/bch709_assembly.fasta --sjdbGTFfile /data/gpfs/assoc/bch709/<YOURID>/genomernaseq/bch709.gtf --runThreadN 2 --genomeSAindexNbases 10
RNA Seq mapping
#!/bin/bash
#SBATCH --job-name=star_wt
#SBATCH --cpus-per-task=2
#SBATCH --time=12:00:00
#SBATCH --mem=8g
#SBATCH --mail-type=all
#SBATCH --mail-user=<EMAIL>@unr.edu
#SBATCH -o star.out # STDOUT
#SBATCH -e star.err # STDERR
#SBATCH -p cpu-s2-core-0
#SBATCH -A cpu-s2-bch709-0
STAR --alignIntronMax 1000000 --alignEndsType EndToEnd --alignTranscriptsPerReadNmax 50000 --runThreadN 2 --outSAMtype BAM SortedByCoordinate --genomeDir /data/gpfs/assoc/bch709/<YOURID>/genomernaseq/reference --readFilesIn /data/gpfs/assoc/bch709/Course_material/2020/RNASeq_trimmed_fastq/WT1_R1_val_1.fq.gz,/data/gpfs/assoc/bch709/Course_material/2020/RNASeq_trimmed_fastq/WT1_R2_val_2.fq.gz --outFileNamePrefix WT1 --readFilesCommand zcat
STAR --alignIntronMax 1000000 --alignEndsType EndToEnd --alignTranscriptsPerReadNmax 50000 --runThreadN 2 --outSAMtype BAM SortedByCoordinate --genomeDir /data/gpfs/assoc/bch709/<YOURID>/genomernaseq/reference --readFilesIn /data/gpfs/assoc/bch709/Course_material/2020/RNASeq_trimmed_fastq/WT2_R1_val_1.fq.gz,/data/gpfs/assoc/bch709/Course_material/2020/RNASeq_trimmed_fastq/WT2_R2_val_2.fq.gz --outFileNamePrefix WT2 --readFilesCommand zcat
STAR --alignIntronMax 1000000 --alignEndsType EndToEnd --alignTranscriptsPerReadNmax 50000 --runThreadN 2 --outSAMtype BAM SortedByCoordinate --genomeDir /data/gpfs/assoc/bch709/<YOURID>/genomernaseq/reference --readFilesIn /data/gpfs/assoc/bch709/Course_material/2020/RNASeq_trimmed_fastq/WT3_R1_val_1.fq.gz,/data/gpfs/assoc/bch709/Course_material/2020/RNASeq_trimmed_fastq/WT3_R2_val_2.fq.gz --outFileNamePrefix WT3 --readFilesCommand zcat
#!/bin/bash
#SBATCH --job-name=star_dt
#SBATCH --cpus-per-task=2
#SBATCH --time=12:00:00
#SBATCH --mem=8g
#SBATCH --mail-type=all
#SBATCH --mail-user=<EMAIL>@unr.edu
#SBATCH -o star.out # STDOUT
#SBATCH -e star.err # STDERR
#SBATCH -p cpu-s2-core-0
#SBATCH -A cpu-s2-bch709-0
STAR --alignIntronMax 1000000 --alignEndsType EndToEnd --alignTranscriptsPerReadNmax 50000 --runThreadN 2 --outSAMtype BAM SortedByCoordinate --genomeDir /data/gpfs/assoc/bch709/<YOURID>/genomernaseq/reference --readFilesIn /data/gpfs/assoc/bch709/Course_material/2020/RNASeq_trimmed_fastq/DT1_R1_val_1.fq.gz,/data/gpfs/assoc/bch709/Course_material/2020/RNASeq_trimmed_fastq/DT1_R2_val_2.fq.gz --outFileNamePrefix DT1 --readFilesCommand zcat
STAR --alignIntronMax 1000000 --alignEndsType EndToEnd --alignTranscriptsPerReadNmax 50000 --runThreadN 2 --outSAMtype BAM SortedByCoordinate --genomeDir /data/gpfs/assoc/bch709/<YOURID>/genomernaseq/reference --readFilesIn /data/gpfs/assoc/bch709/Course_material/2020/RNASeq_trimmed_fastq/DT2_R1_val_1.fq.gz,/data/gpfs/assoc/bch709/Course_material/2020/RNASeq_trimmed_fastq/DT2_R2_val_2.fq.gz --outFileNamePrefix DT2 --readFilesCommand zcat
STAR --alignIntronMax 1000000 --alignEndsType EndToEnd --alignTranscriptsPerReadNmax 50000 --runThreadN 2 --outSAMtype BAM SortedByCoordinate --genomeDir /data/gpfs/assoc/bch709/<YOURID>/genomernaseq/reference --readFilesIn /data/gpfs/assoc/bch709/Course_material/2020/RNASeq_trimmed_fastq/DT3_R1_val_1.fq.gz,/data/gpfs/assoc/bch709/Course_material/2020/RNASeq_trimmed_fastq/DT3_R2_val_2.fq.gz --outFileNamePrefix DT3 --readFilesCommand zcat
Reads count
In the case of RNA-Seq, the features are typically genes, where each gene is considered here as the union of all its exons. Counting RNA-seq reads is complex because of the need to accommodate exon splicing. The common approach is to summarize counts at the gene level, by counting all reads that overlap any exon for each gene. In this method, gene annotation file from RefSeq or Ensembl is often used for this purpose. So far there are two major feature counting tools: featureCounts (Liao et al.) and htseq-count (Anders et al.)

#!/bin/bash
#SBATCH --job-name=featureCounts
#SBATCH --cpus-per-task=16
#SBATCH --time=15:00
#SBATCH --mem=20g
#SBATCH --mail-type=all
#SBATCH --mail-user=<EMAIL>@unr.edu
#SBATCH -o featureCounts.out # STDOUT
#SBATCH -e featureCounts.err # STDERR
#SBATCH -p cpu-s6-test-0
#SBATCH -A cpu-s6-test-0
featureCounts -Q 10 -M -s 0 -T 16 -p -a bch709.gtf WT1Aligned.sortedByCoord.out.bam WT2Aligned.sortedByCoord.out.bam WT3Aligned.sortedByCoord.out.bam DT1Aligned.sortedByCoord.out.bam DT2Aligned.sortedByCoord.out.bam DT3Aligned.sortedByCoord.out.bam -o BCH709.featureCount.cnt
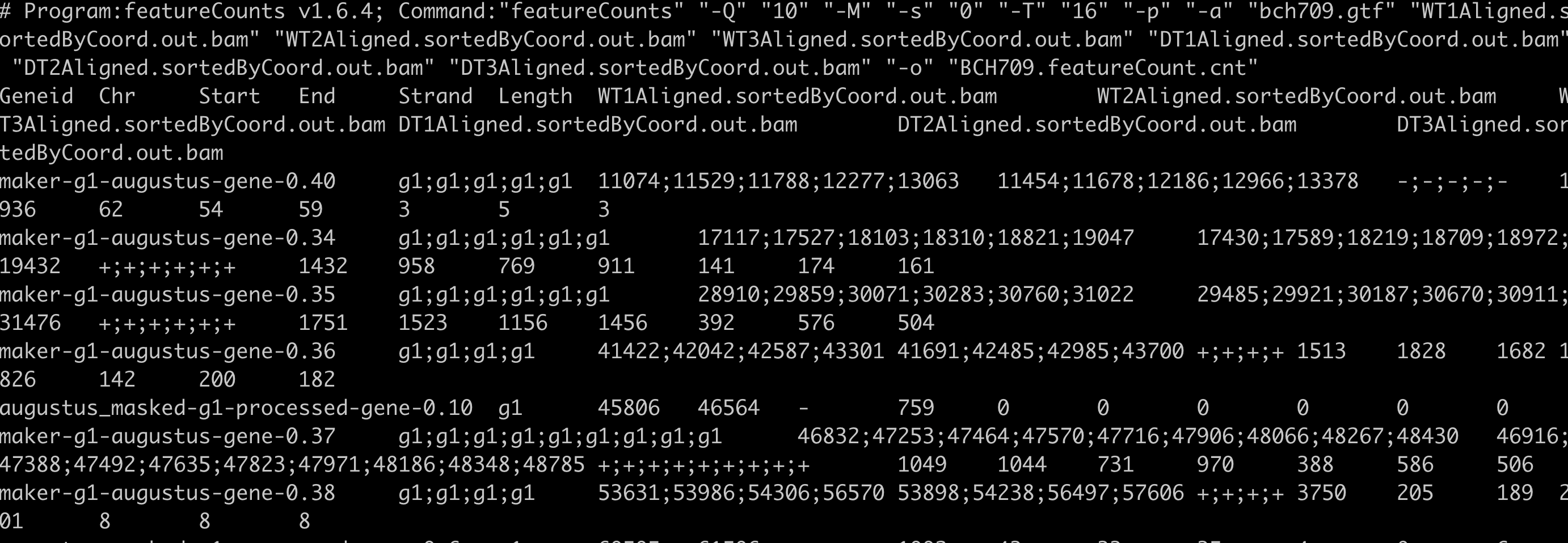
cut -f1,7- BCH709.featureCount.cnt | egrep -v "#" | sed 's/\Aligned\.sortedByCoord\.out\.bam//g' >> BCH709.featureCount_count_only.cnt
sample files
nano samples.txt
WT<TAB>WT1
WT<TAB>WT2
WT<TAB>WT3
DT<TAB>DT1
DT<TAB>DT2
DT<TAB>DT3
PtR (Quality Check Your Samples and Biological Replicates)
Once you’ve performed transcript quantification for each of your biological replicates, it’s good to examine the data to ensure that your biological replicates are well correlated, and also to investigate relationships among your samples. If there are any obvious discrepancies among your sample and replicate relationships such as due to accidental mis-labeling of sample replicates, or strong outliers or batch effects, you’ll want to identify them before proceeding to subsequent data analyses (such as differential expression).
PtR --matrix BCH709.featureCount_count_only.cnt --samples samples.txt --CPM --log2 --min_rowSums 10 --sample_cor_matrix --compare_replicates
WT.rep_compare.pdf
DT.rep_compare.pdf
DEG calculation
run_DE_analysis.pl --matrix BCH709.featureCount_count_only.cnt --method DESeq2 --samples_file samples.txt --output rnaseq
TPM and FPKM calculation
cut -f1,6- BCH709.featureCount.cnt | egrep -v "#" | sed 's/\Aligned\.sortedByCoord\.out\.bam//g' >> BCH709.featureCount_count_length.cnt
python /data/gpfs/assoc/bch709/Course_material/2020/script/tpm_raw_exp_calculator.py -count BCH709.featureCount_count_length.cnt
TPM and FPKM calculation output
BCH709.featureCount_count_length.cnt.fpkm.xls
BCH709.featureCount_count_length.cnt.fpkm.tab
BCH709.featureCount_count_length.cnt.tpm.xls
BCH709.featureCount_count_length.cnt.tpm.tab
DEG subset
cd rnaseq
analyze_diff_expr.pl --samples ../samples.txt --matrix ../BCH709.featureCount_count_length.cnt.tpm.tab