Quick Reminder
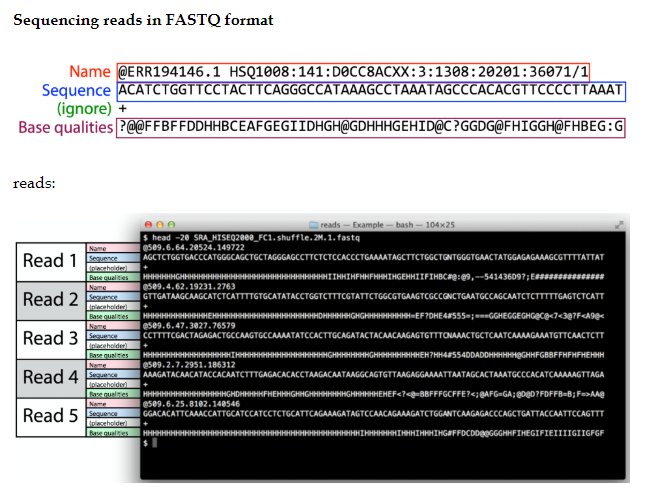

RNASeq
$ cd /data/gpfs/assoc/bch709/YOURID
$ mkdir rnaseq/
$ cd rnaseq
$ conda activate rnaseq
File copy
$ cd /data/gpfs/assoc/bch709/YOURID/
$ cd rnaseq/transcriptome_assembly/fastq
$ cp /data/gpfs/assoc/bch709/Course_material/2020/RNASeq/*.gz .
FASTQC
$ pwd
/data/gpfs/assoc/bch709/YOURID/rnaseq/transcriptome_assembly/fastq
$ conda install -c bioconda fastqc
$ fastqc pair1.fastq.gz pair2.fastq.gz
Trim the reads
- Trim IF necessary
- Synthetic bases can be an issue for SNP calling
- Insert size distribution may be more important for assemblers
- Trim/Clip/Filter reads
- Remove adapter sequences
- Trim reads by quality
- Sliding window trimming
- Filter by min/max read length
- Remove reads less than ~18nt
- Demultiplexing/Splitting
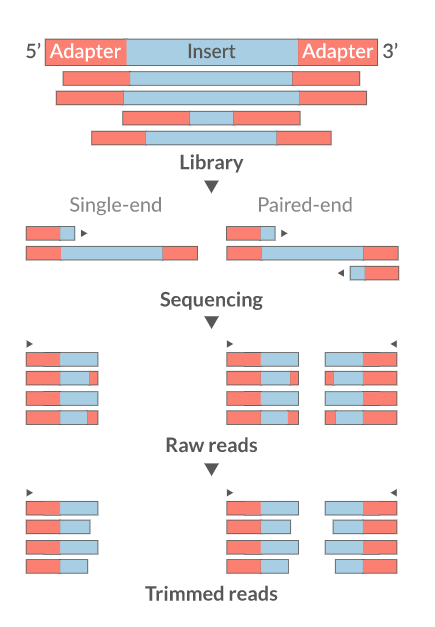
Cutadapt
fastp
Skewer
Prinseq
Trimmomatics
Trim Galore
Install Trim Galore
$ conda install trim-galore
Run trimming
$ trim_galore --help
$ trim_galore --paired --three_prime_clip_R1 20 --three_prime_clip_R2 20 --cores 2 --max_n 40 --gzip -o trim paired1.fastq.gz paired2.fastq.gz
$ ls trim/
Align the reads (mapping)
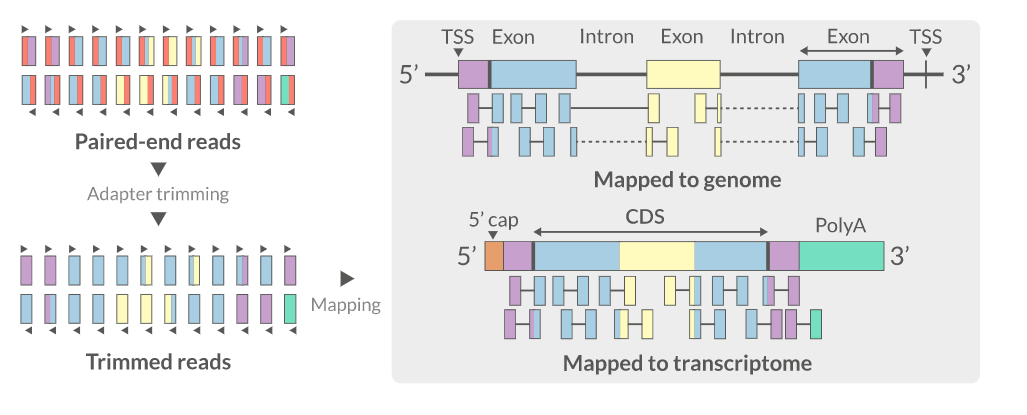
- Aligning reads back to a reference sequence
- Mapping to genome vs transcriptome
- Splice-aware alignment (genome)
STAR
HISAT2
GSNAP
Bowtie2
Novoalign
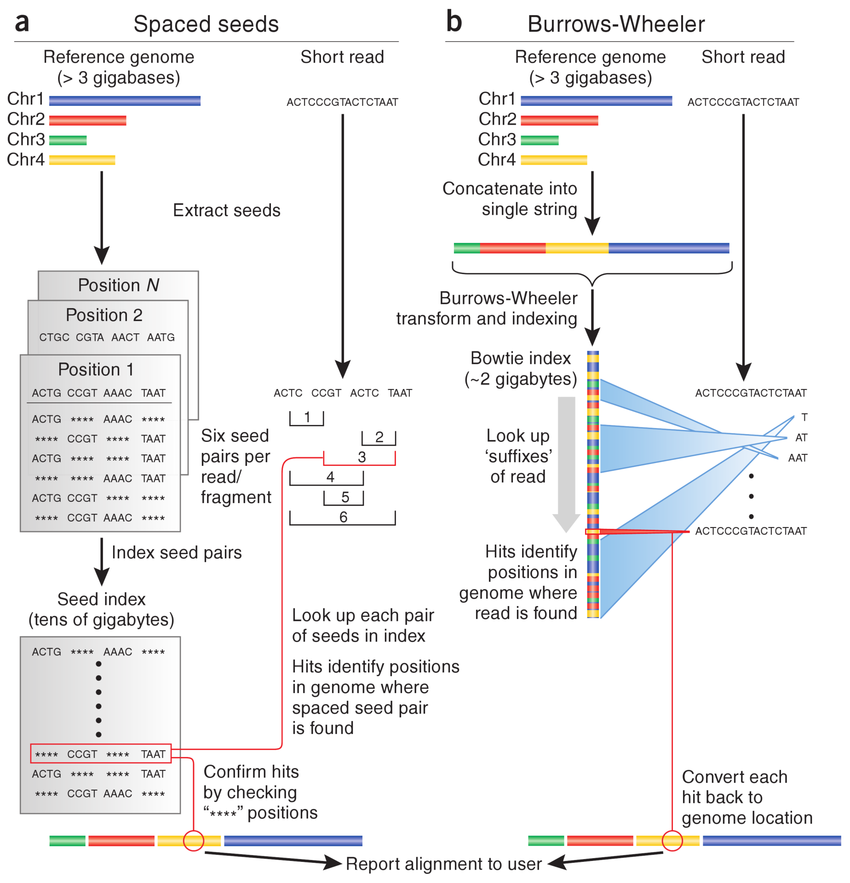

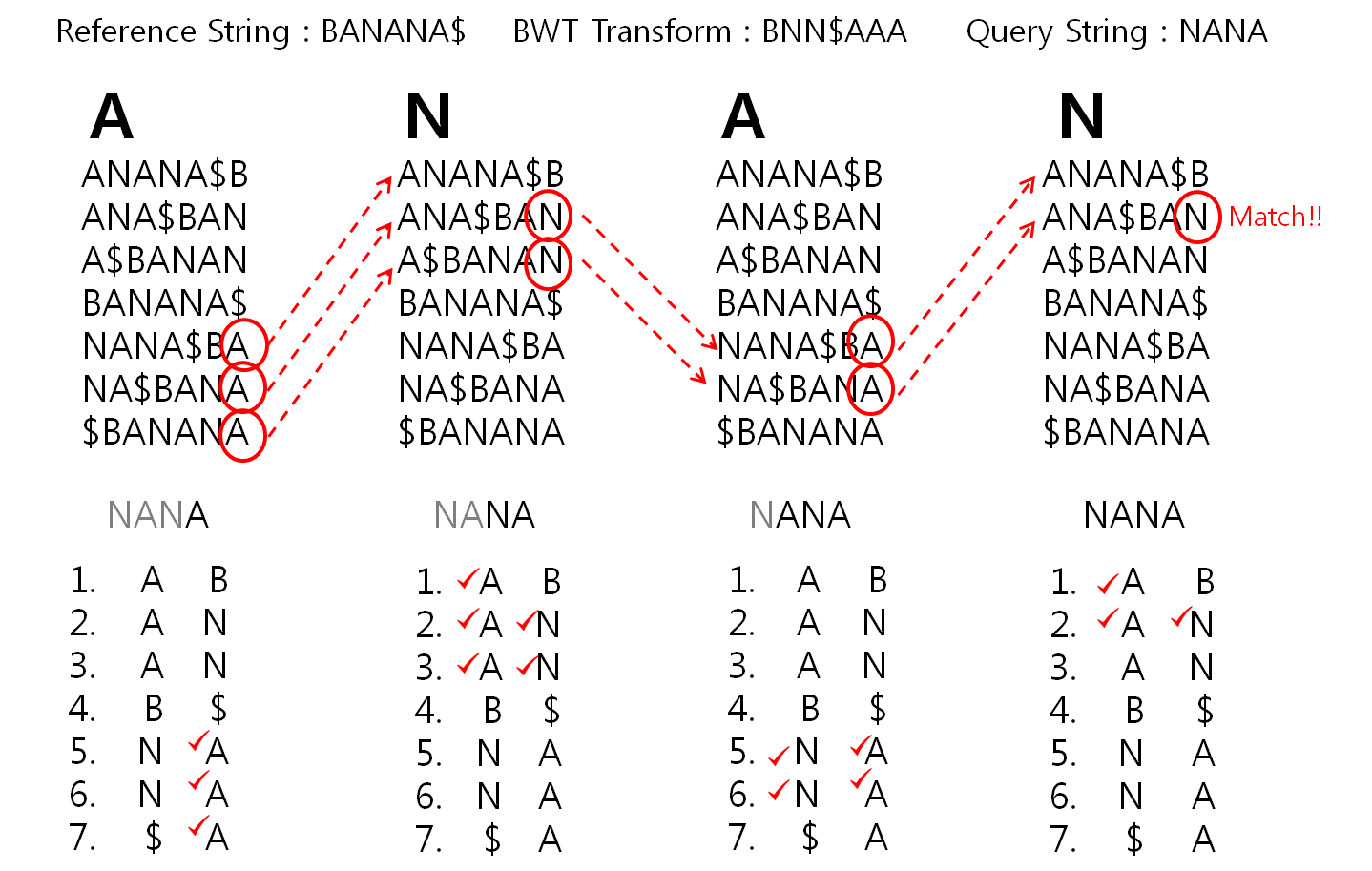
Install HISAT2 (graph FM index, spin off version Burrows-Wheeler Transform)
$ conda install hisat2
Download reference sequence
$ wget https://www.dropbox.com/s/0onch14nnxx9b94/bch709.fasta
HISAT2 indexing
$ hisat2-build --help
$ hisat2-build bch709.fasta bch709
HISAT2 mapping
$ hisat2 -x bch709 --threads <YOUR CPU COUNT> -1 trim/paired1_val_1.fq.gz -2 trim/paired2_val_2.fq.gz -S align.sam
SAM file format
Check result
$ head align.sam
SAM file
<QNAME> <FLAG> <RNAME> <POS> <MAPQ> <CIGAR> <MRNM> <MPOS> <ISIZE> <SEQ> <QUAL> [<TAG>:<VTYPE>:<VALUE> [...]]
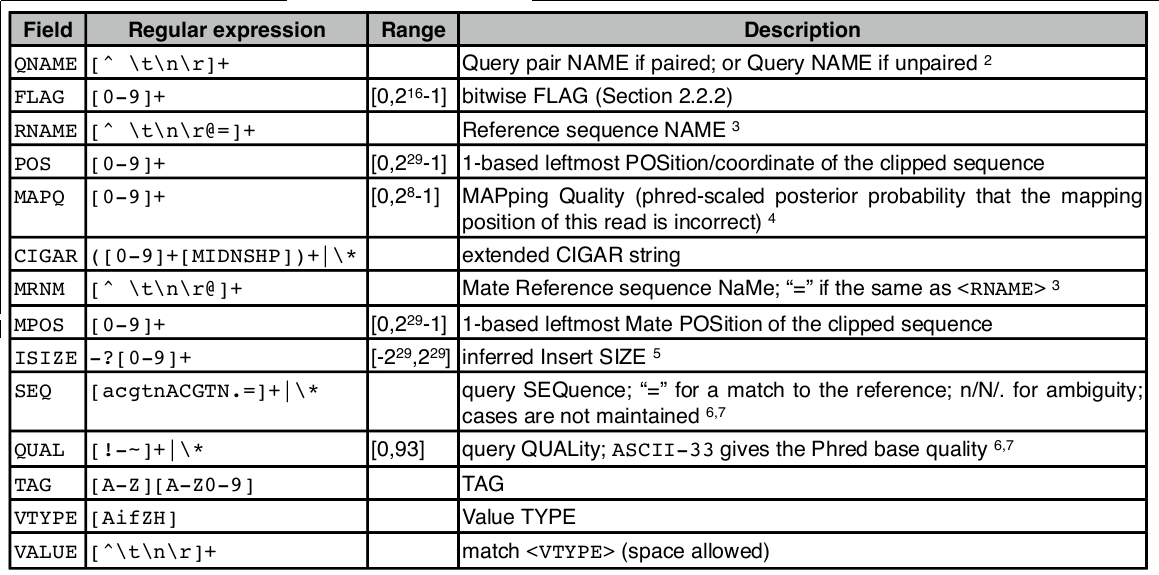
SAM flag

Demical to binary
$ echo 'obase=2;163' | bc
If you don’t have bc, please install through conda
$ conda install bc
SAM tag
There are a bunch of predefined tags, please see the SAM manual for more information. For the tags used in this example:
Any tags that start with X? are reserved fields for end users: XT:A:M, XN:i:2, XM:i:0, XO:i:0, XG:i:0
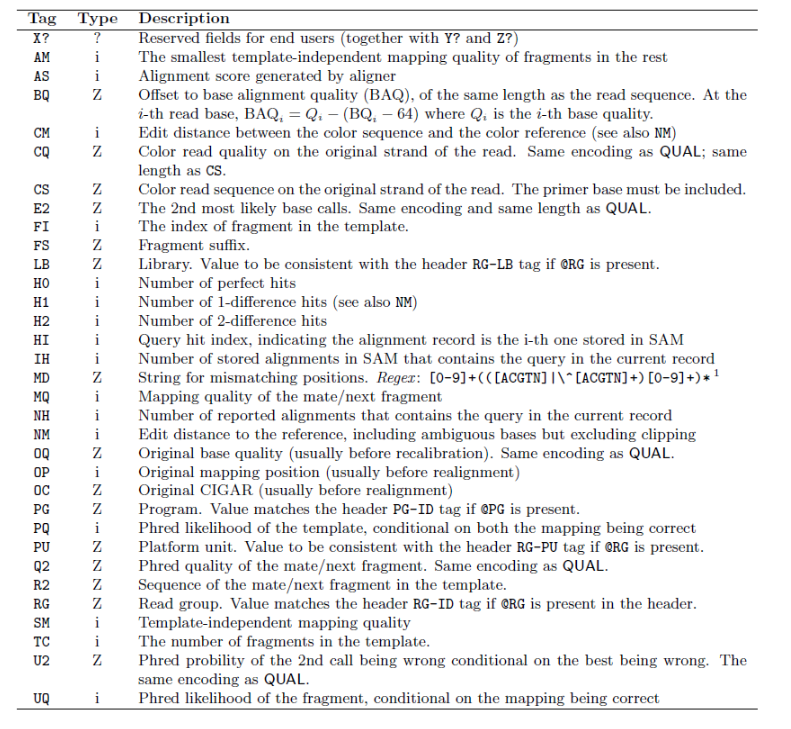
More information is below.
http://samtools.github.io/hts-specs/
SAMtools
SAM (Sequence Alignment/Map) format is a generic format for storing large nucleotide sequence alignments. SAM aims to be a format that:
- Is flexible enough to store all the alignment information generated by various alignment programs;
- Is simple enough to be easily generated by alignment programs or converted from existing alignment formats;
- Is compact in file size;
- Allows most of operations on the alignment to work on a stream without loading the whole alignment into memory;
- Allows the file to be indexed by genomic position to efficiently retrieve all reads aligning to a locus.
SAM Tools provide various utilities for manipulating alignments in the SAM format, including sorting, merging, indexing and generating alignments in a per-position format. http://samtools.sourceforge.net/
$ samtools view -Sb align.sam > align.bam
$ samtools sort align.bam -o align_sort.bam
$ samtools index align_sort.bam
BAM file
A BAM file (.bam) is the binary version of a SAM file. A SAM file (.sam) is a tab-delimited text file that contains sequence alignment data.
| SAM/BAM | size |
|---|---|
| align.sam | 903M |
| align.bam | 166M |
Alignment visualization
samtools tview align_sort.bam bch709.fasta
How to make a report?

$ conda install 'multiqc<1.34'
$ multiqc --help
$ multiqc . --exclude rsem --exclude gatk
Alignment QC
- Number of reads mapped/unmapped/paired etc
- Uniquely mapped
- Insert size distribution
- Coverage
- Gene body coverage
- Biotype counts / Chromosome counts
- Counts by region: gene/intron/non-genic
- Sequencing saturation
- Strand specificity
samtools > stats
bamtools > stats
QoRTs
RSeQC
Qualimap
Quantification • Counts
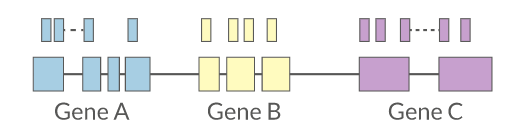
- Read counts = gene expression
- Reads can be quantified on any feature (gene, transcript, exon etc)
- Intersection on gene models
- Gene/Transcript level

featureCounts HTSeq RSEM Cufflinks Rcount
conda install -c bioconda subread
conda install -c bioconda rsem
Quantification method
- PCR duplicates
- Ignore for RNA-Seq data
- Computational deduplication (Don’t!)
- Use PCR-free library-prep kits
-
Use UMIs during library-prep
- Multi-mapping
- Added (BEDTools multicov)
- Discard (featureCounts, HTSeq)
- Distribute counts (Cufflinks)
- Rescue
- Probabilistic assignment (Rcount, Cufflinks)
- Prioritise features (Rcount)
- Probabilistic assignment with EM (RSEM)
Reference
Fu, Yu, et al. “Elimination of PCR duplicates in RNA-seq and small RNA-seq using unique molecular identifiers.” BMC genomics 19.1 (2018): 531 Parekh, Swati, et al. “The impact of amplification on differential expression analyses by RNA-seq.” Scientific reports 6 (2016): 25533 Klepikova, Anna V., et al. “Effect of method of deduplication on estimation of differential gene expression using RNA-seq.” PeerJ 5 (2017): e3091
Quick reminder
conda activate rnaseq
conda install python=3 jellyfish bowtie2 salmon cmake htop
fastqc paired1.fastq.gz paired2.fastq.gz
trim_galore --clip_R1 10 --clip_R2 10 --paired --three_prime_clip_R1 20 --three_prime_clip_R2 20 --cores 2 --max_n 40 --gzip -o trim/ paired1.fastq.gz paired2.fastq.gz
fastqc trim/paired1_val_1.fq.gz trim/paired2_val_2.fq.gz
hisat2-build bch709.fasta bch709
hisat2 -x bch709 --threads 8 -1 trim/paired1_val_1.fq.gz -2 trim/paired2_val_2.fq.gz -S align.sam
samtools view -Sb align.sam > align.bam
samtools sort align.bam -o align_sort.bam
samtools index align_sort.bam
samtools tview align_sort.bam
Transcriptome Assembly
De novo assembly
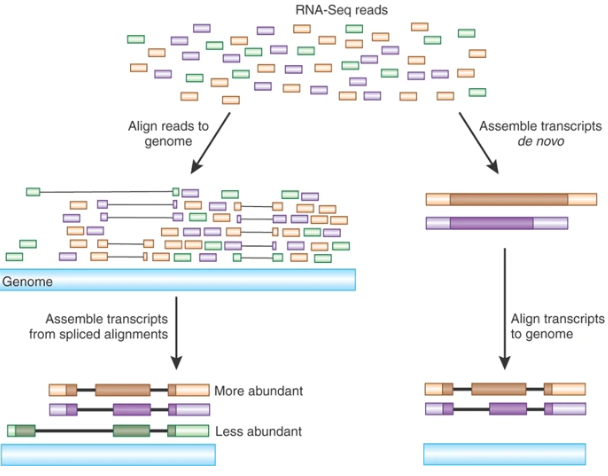
Assembly?


Sequencing coverage


Assembly law
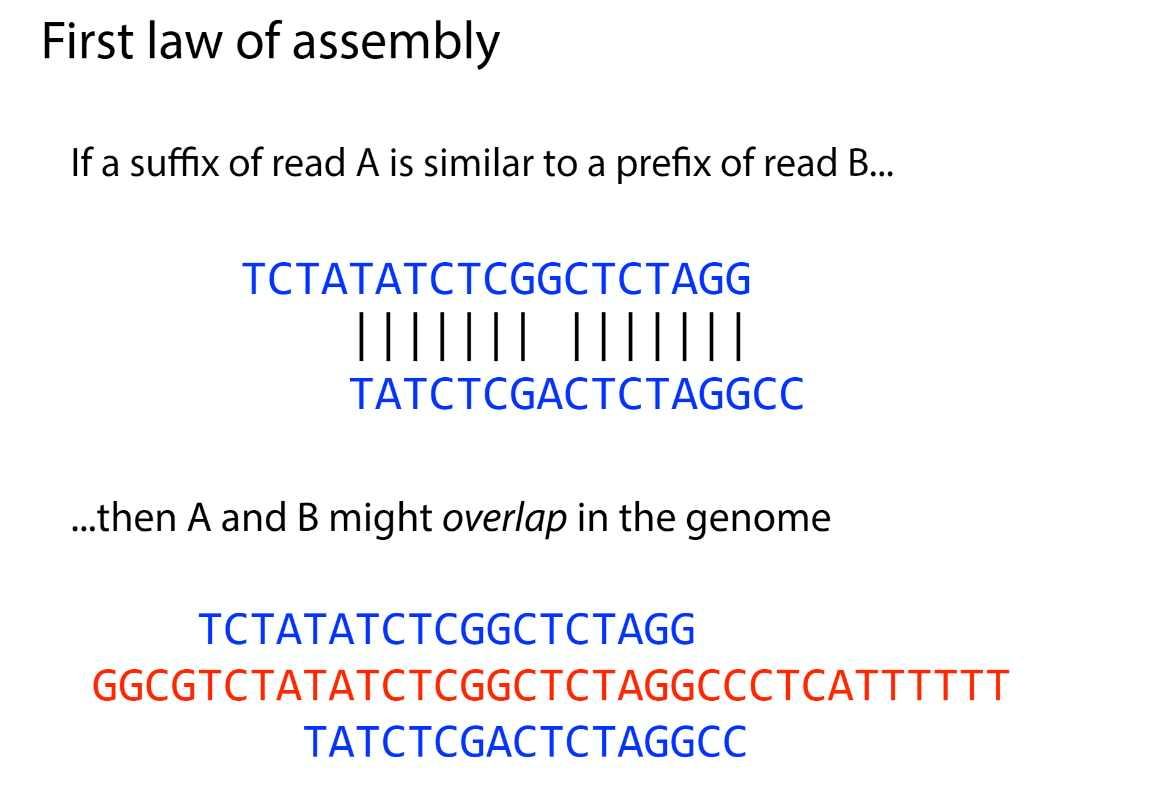
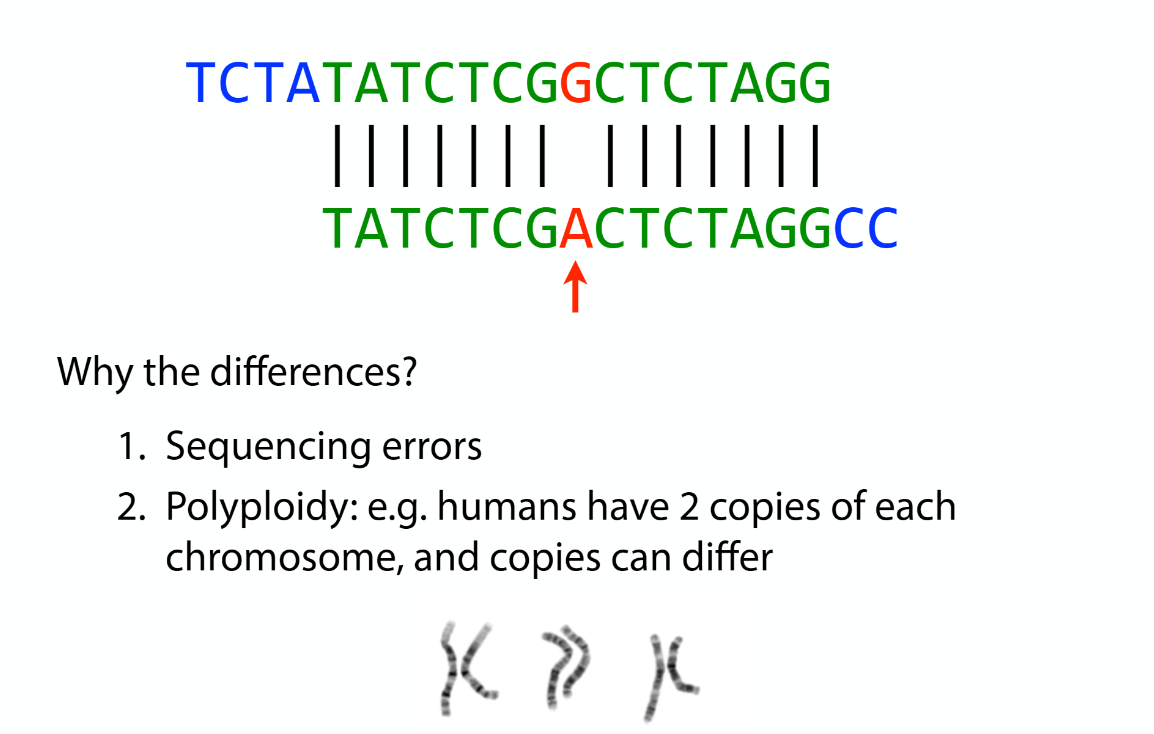
Overlap graph
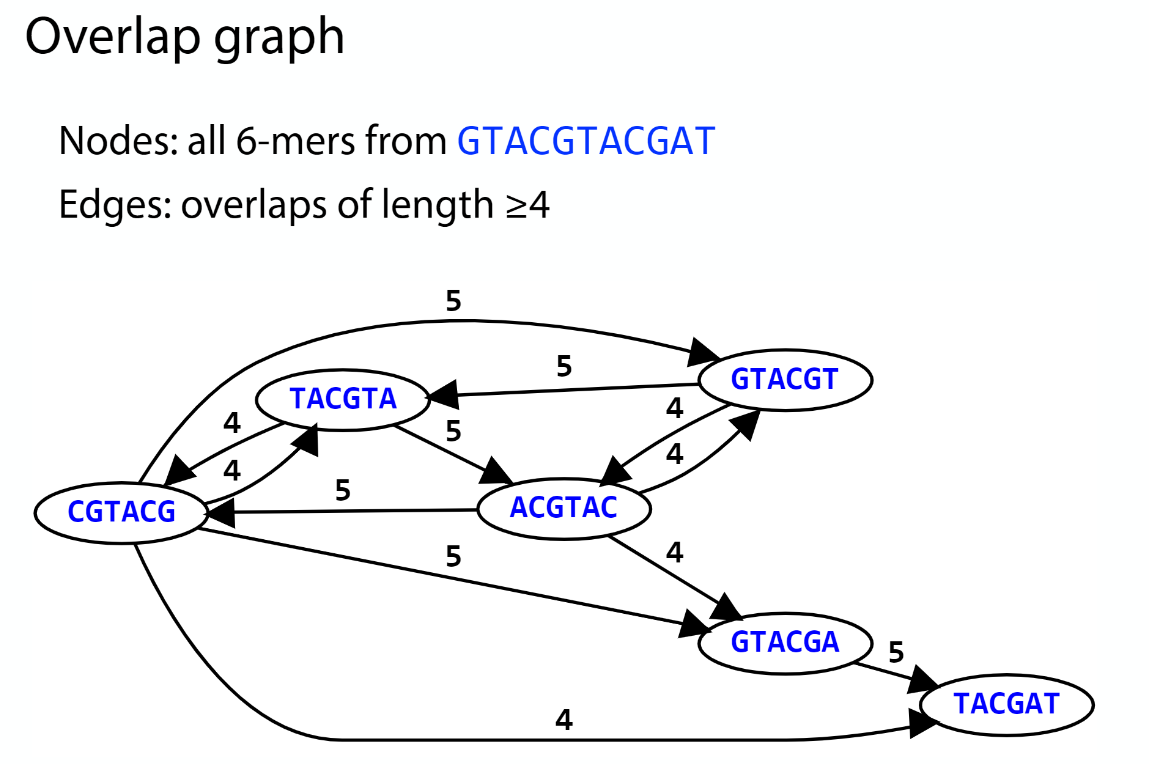
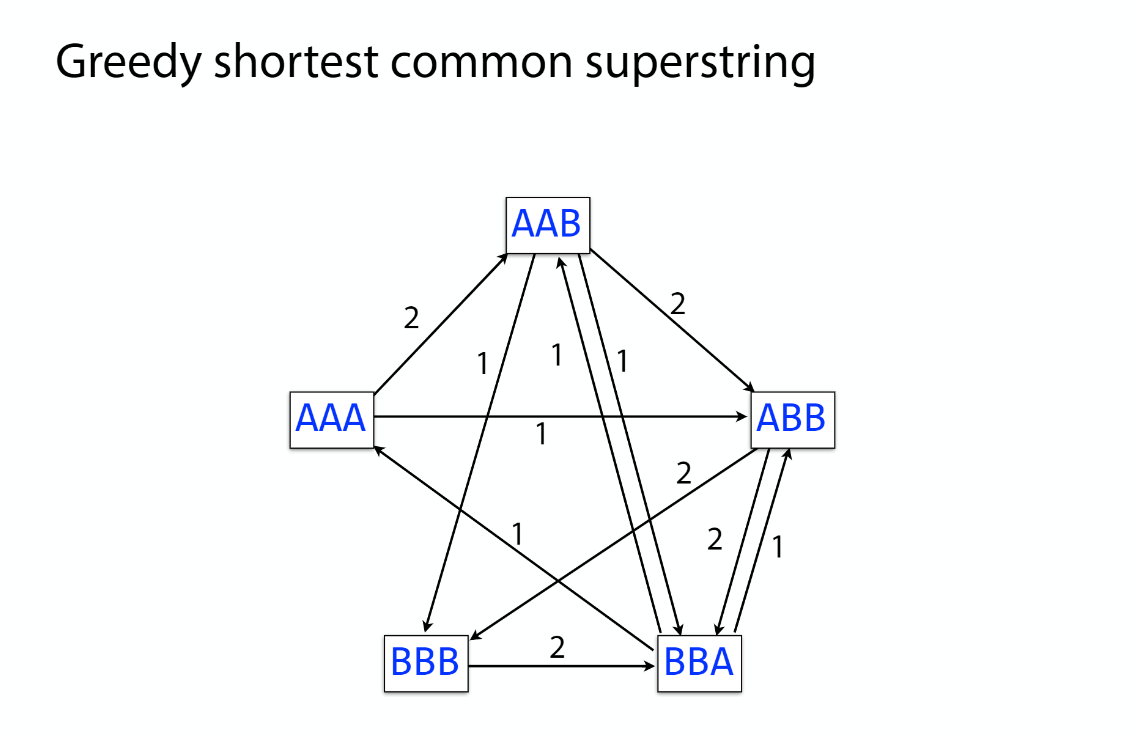
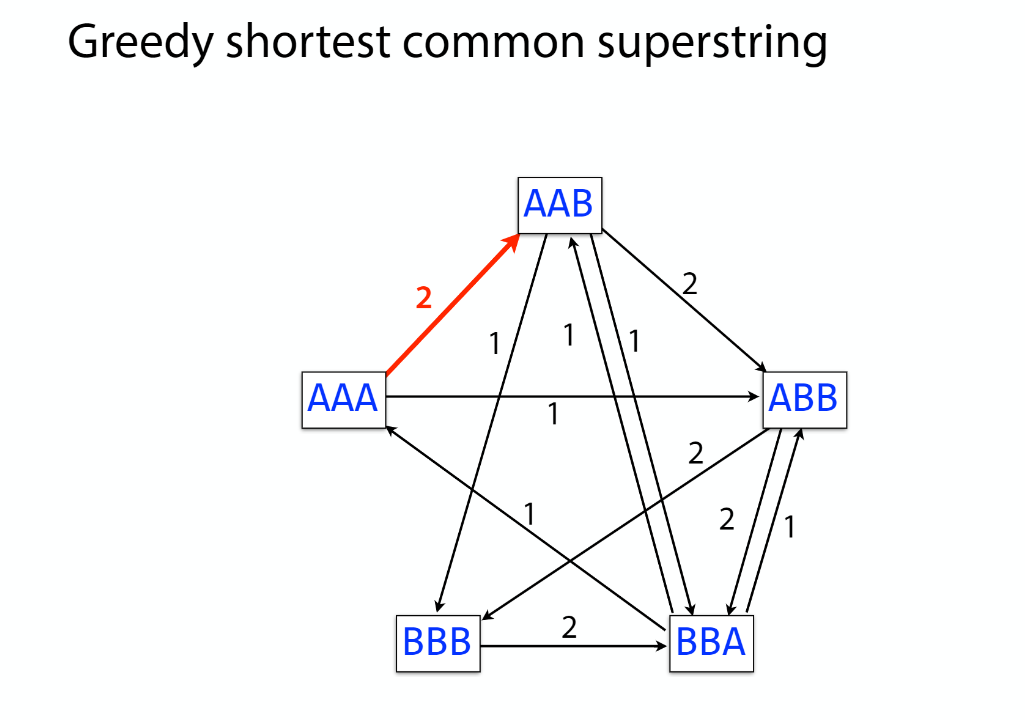
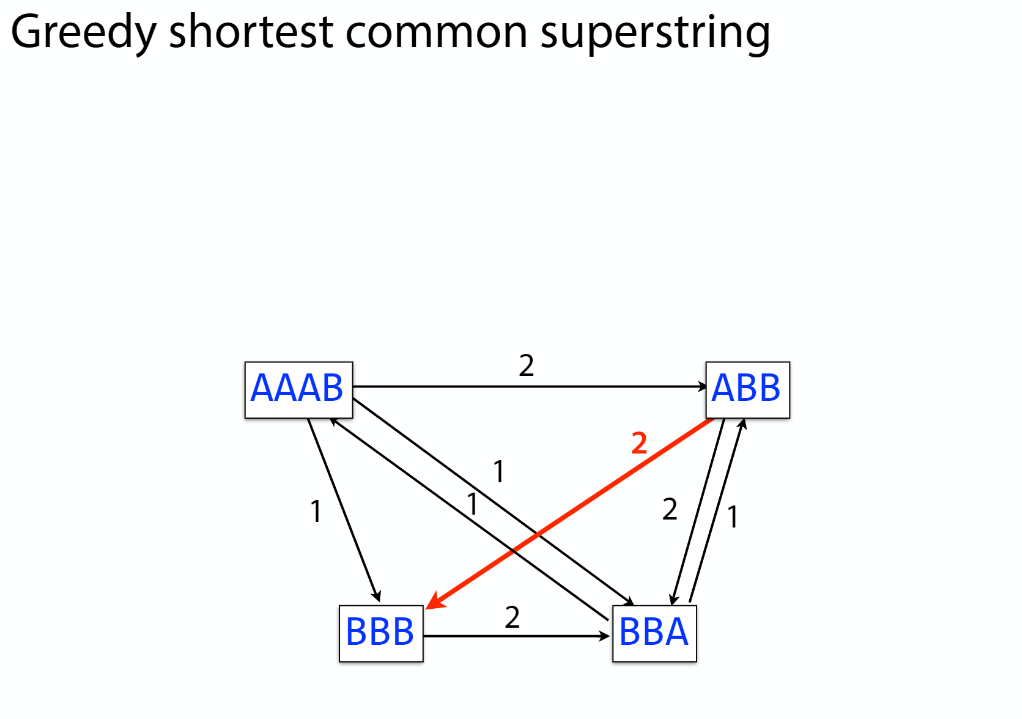
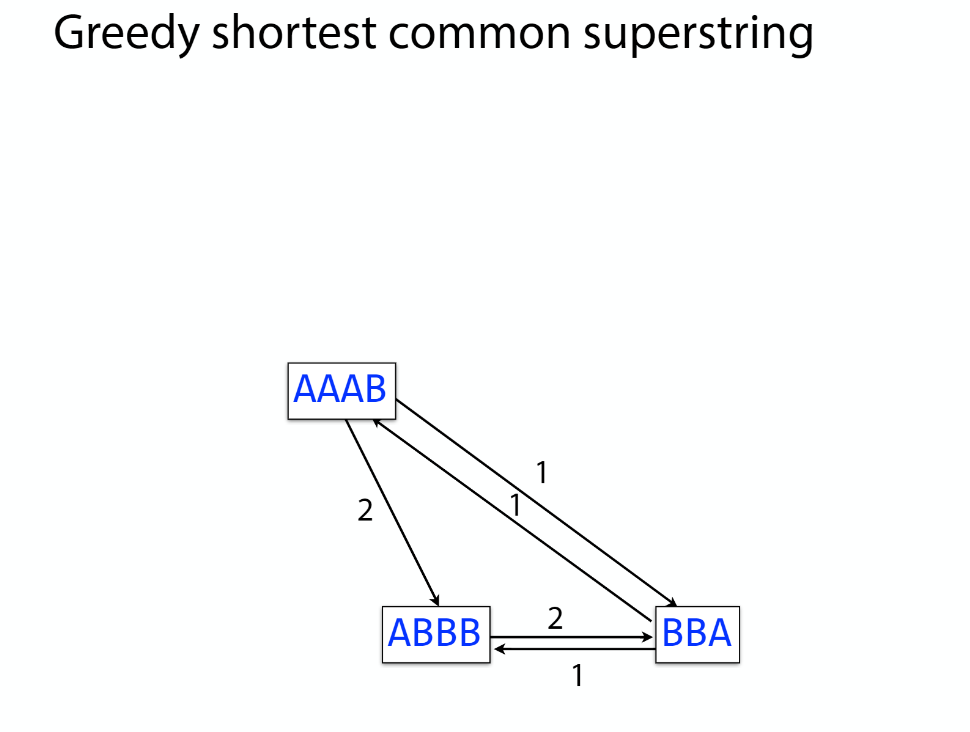
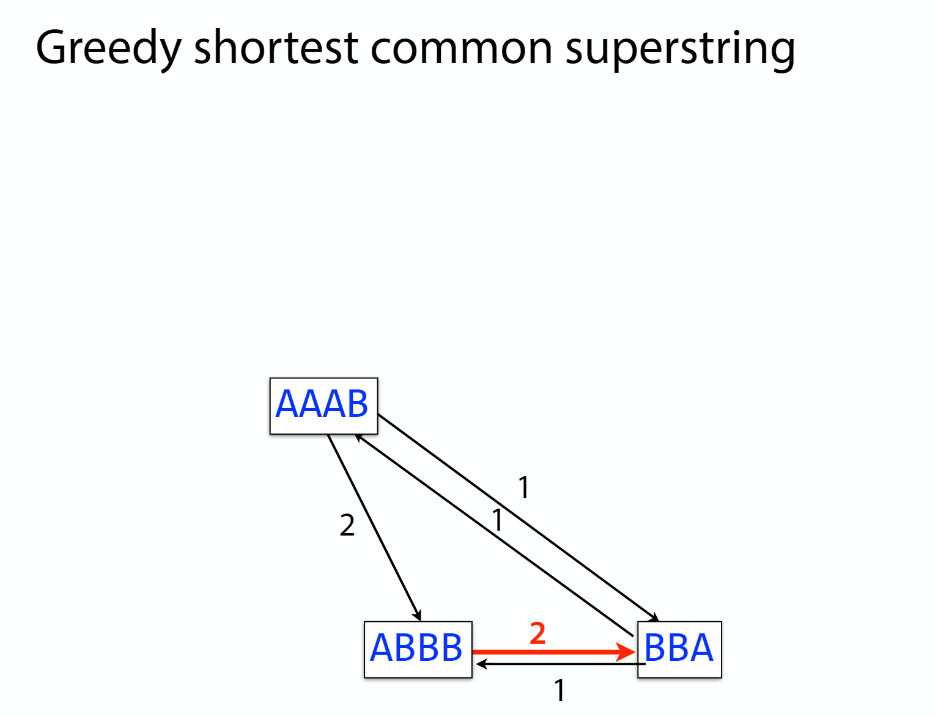
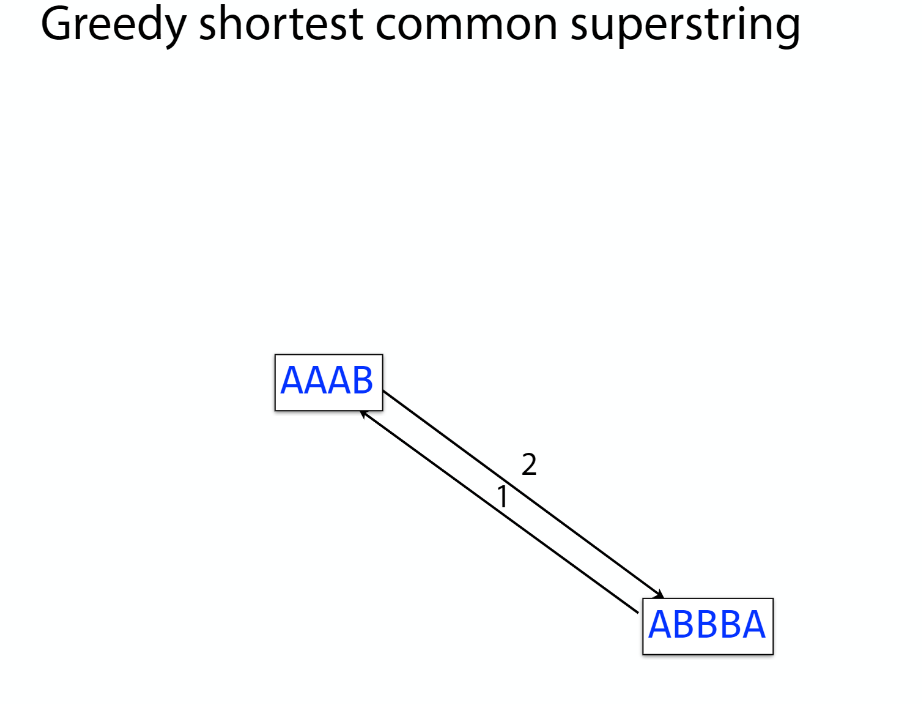
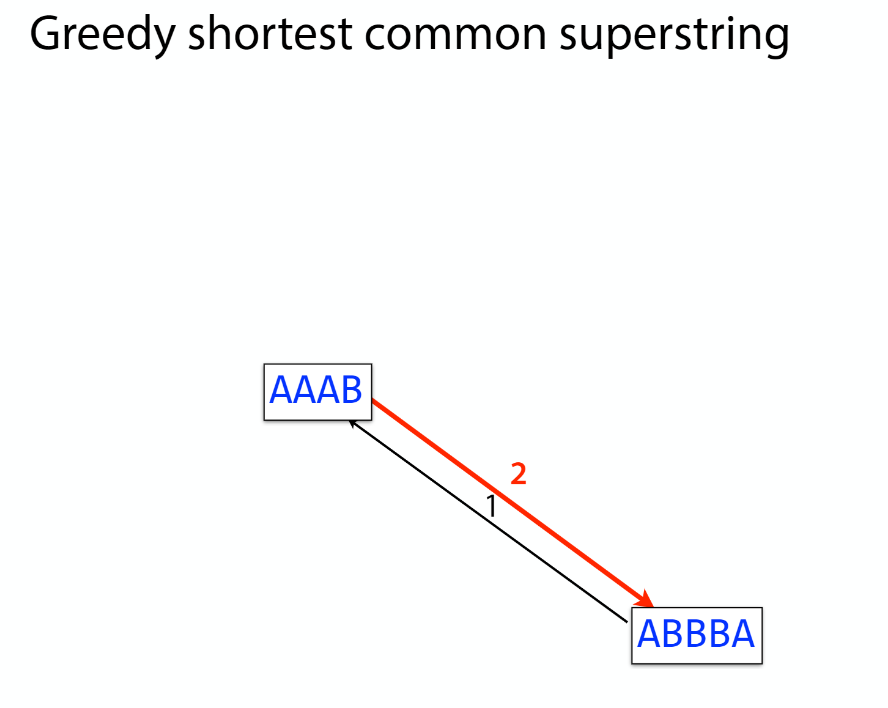
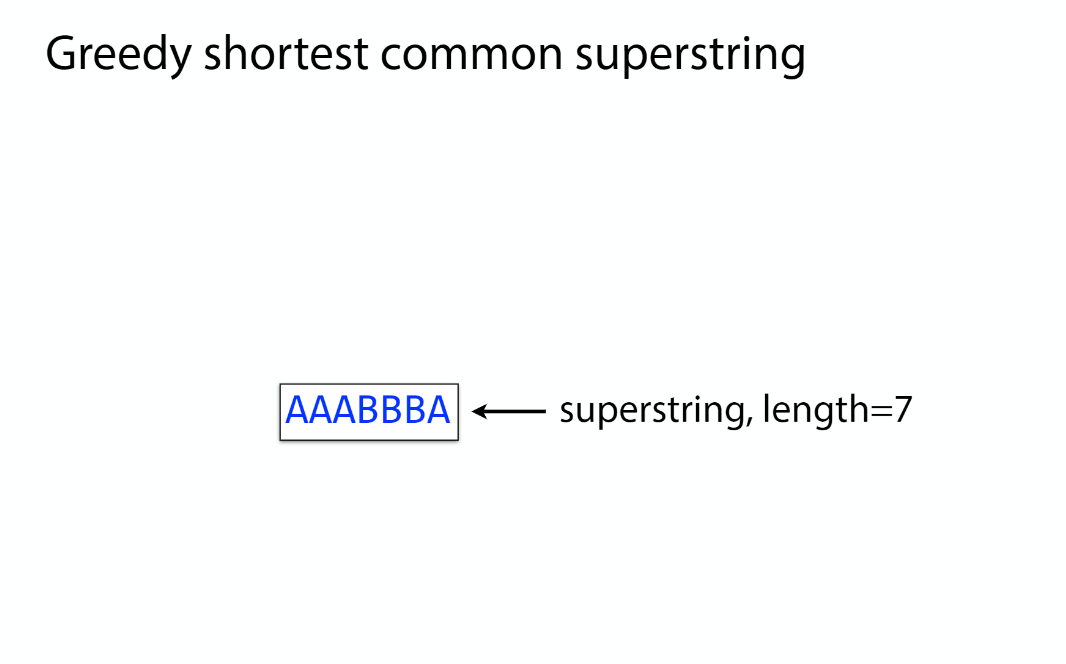

K-mer: substring of length k
k-mers are subsequences of length k contained within a biological sequence.

De bruijn
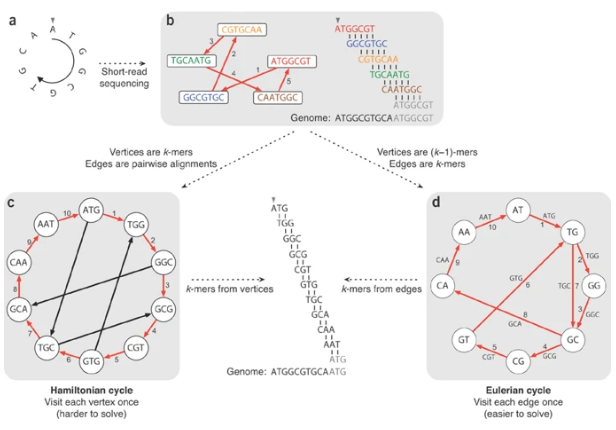
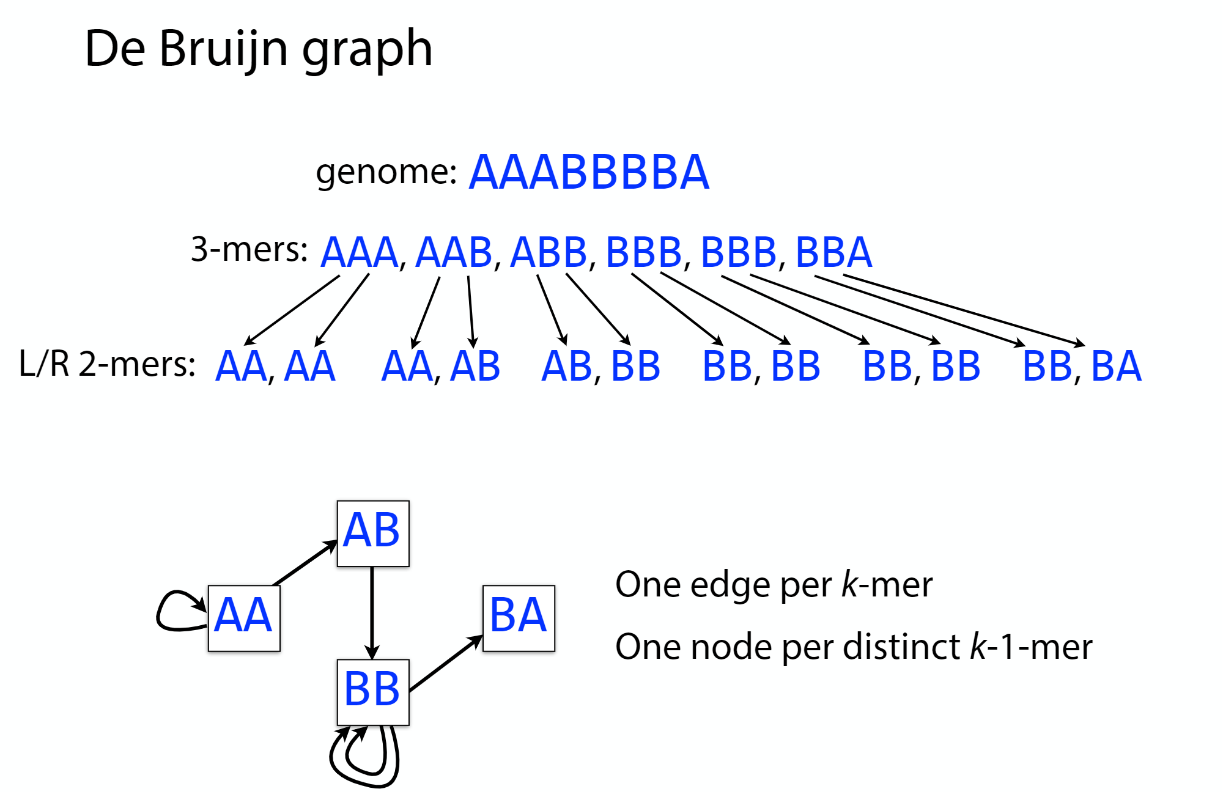
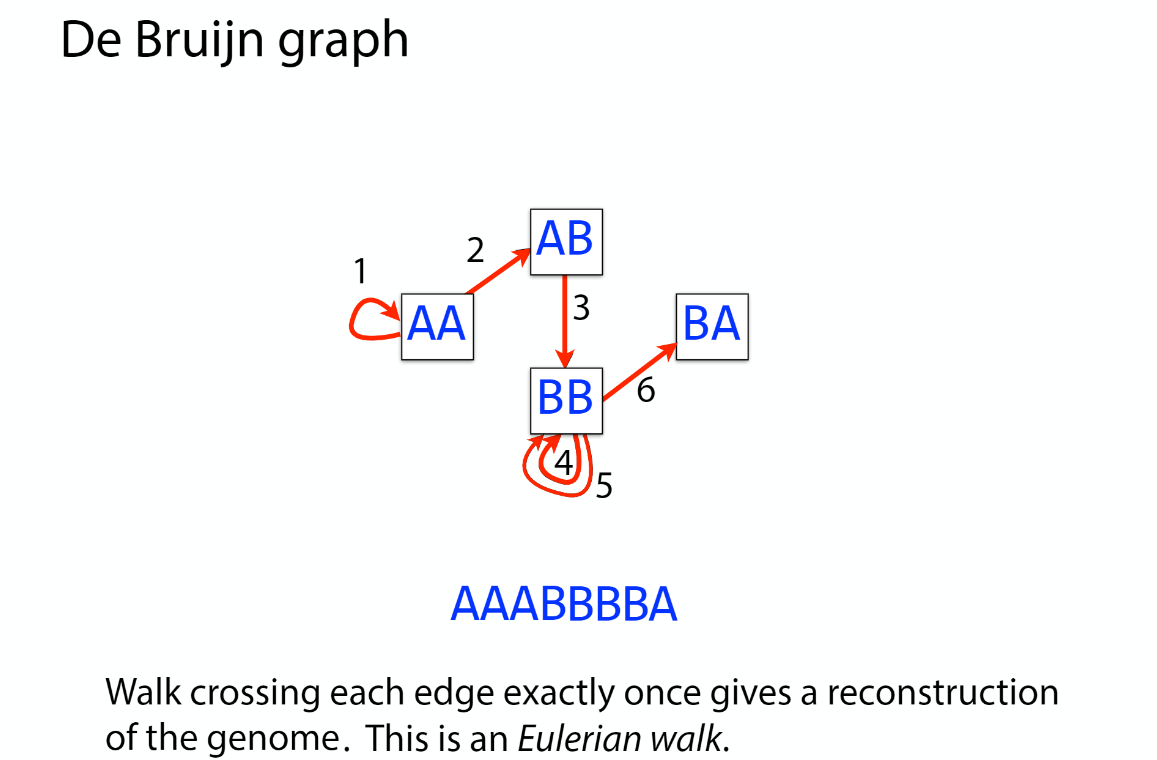
OLC vs De bruijn
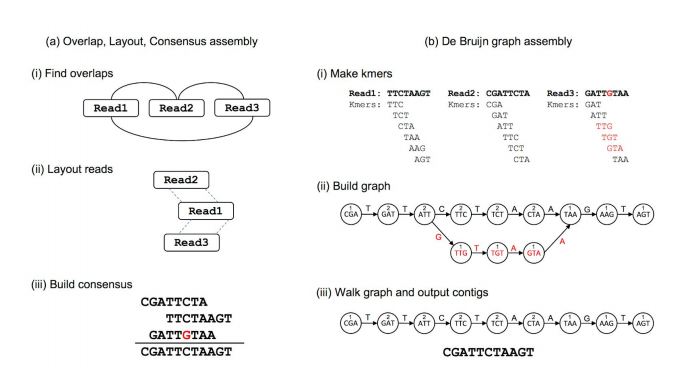
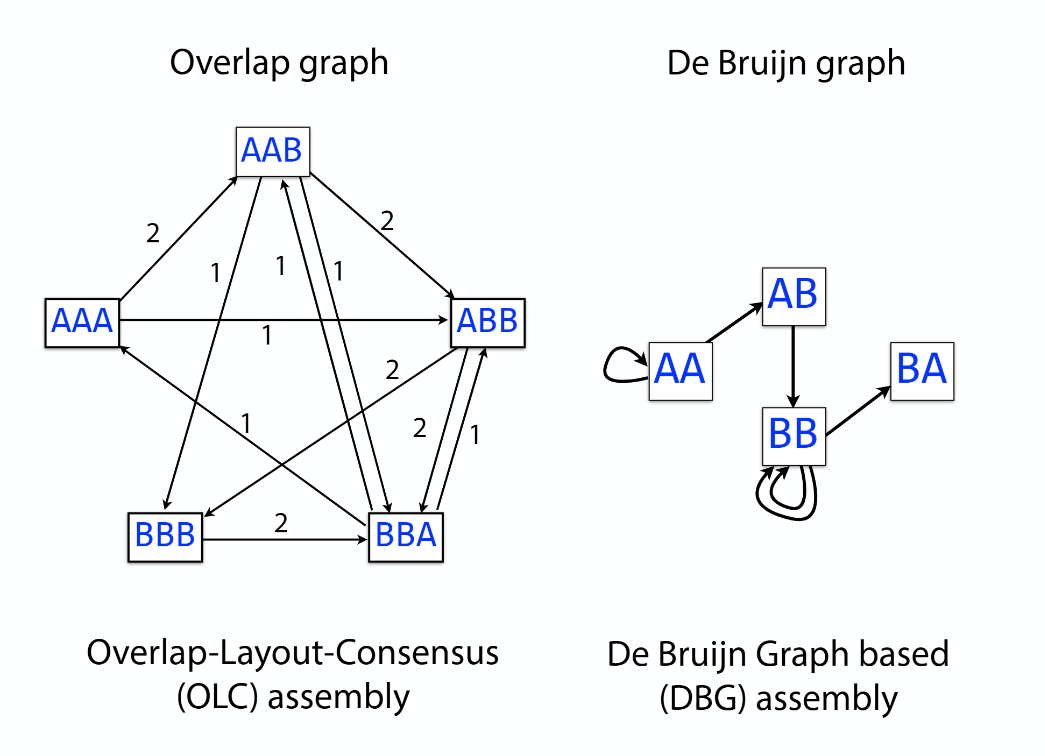

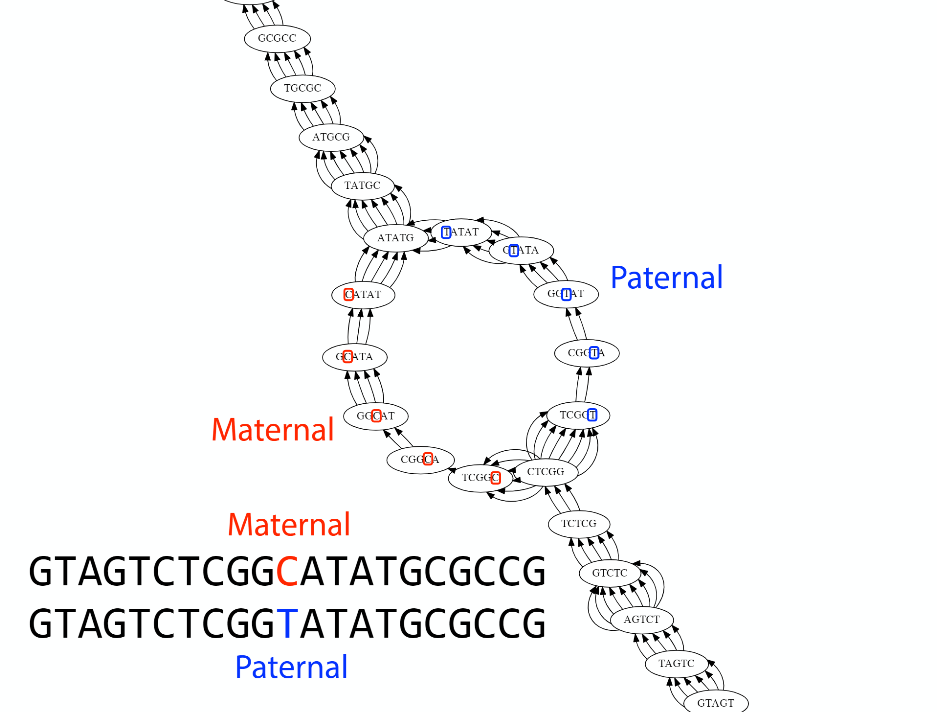
Transcriptome assembler
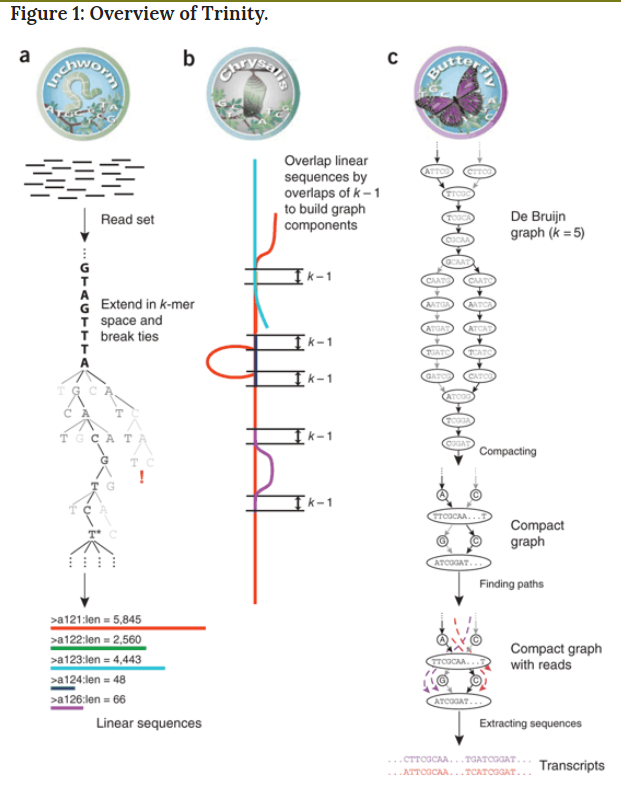
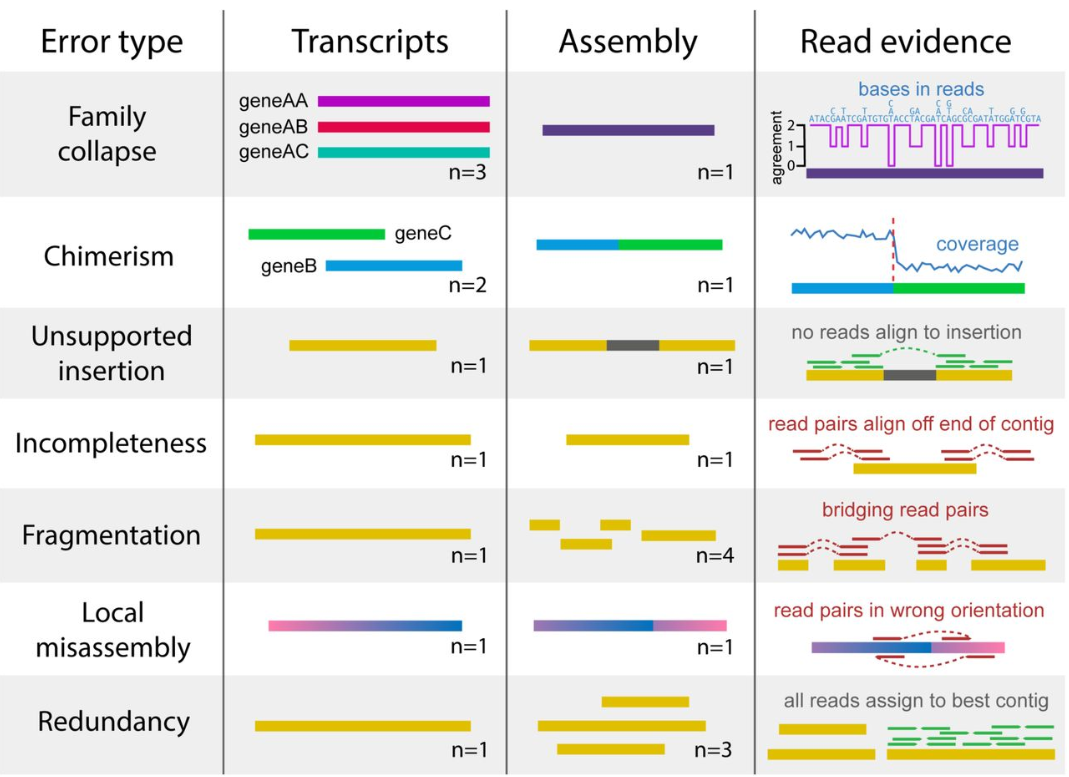
Transcriptome assembly error
Running Trinity
Trinity is run via the script: ‘Trinity’ found in the base installation directory.
Usage info is as follows:
conda create -n transcriptome_assembly
conda activate transcriptome_assembly
conda install -c bioconda/label/cf201901 trinity
###############################################################################
#
# ______ ____ ____ ____ ____ ______ __ __
# | || \ | || \ | || || | |
# | || D ) | | | _ | | | | || | |
# |_| |_|| / | | | | | | | |_| |_|| ~ |
# | | | \ | | | | | | | | | |___, |
# | | | . \ | | | | | | | | | | |
# |__| |__|\_||____||__|__||____| |__| |____/
#
###############################################################################
#
# Required:
#
# --seqType <string> :type of reads: ( fa, or fq )
#
# --max_memory <string> :suggested max memory to use by Trinity where limiting can be enabled. (jellyfish, sorting, etc)
# provided in Gb of RAM, ie. '--max_memory 10G'
#
# If paired reads:
# --left <string> :left reads, one or more file names (separated by commas, not spaces)
# --right <string> :right reads, one or more file names (separated by commas, not spaces)
#
# Or, if unpaired reads:
# --single <string> :single reads, one or more file names, comma-delimited (note, if single file contains pairs, can use flag: --run_as_paired )
#
# Or,
# --samples_file <string> tab-delimited text file indicating biological replicate relationships.
# ex.
# cond_A cond_A_rep1 A_rep1_left.fq A_rep1_right.fq
# cond_A cond_A_rep2 A_rep2_left.fq A_rep2_right.fq
# cond_B cond_B_rep1 B_rep1_left.fq B_rep1_right.fq
# cond_B cond_B_rep2 B_rep2_left.fq B_rep2_right.fq
#
# # if single-end instead of paired-end, then leave the 4th column above empty.
#
####################################
## Misc: #########################
#
# --SS_lib_type <string> :Strand-specific RNA-Seq read orientation.
# if paired: RF or FR,
# if single: F or R. (dUTP method = RF)
# See web documentation.
#
# --CPU <int> :number of CPUs to use, default: 2
# --min_contig_length <int> :minimum assembled contig length to report
# (def=200)
#
# --long_reads <string> :fasta file containing error-corrected or circular consensus (CCS) pac bio reads
#
# --genome_guided_bam <string> :genome guided mode, provide path to coordinate-sorted bam file.
# (see genome-guided param section under --show_full_usage_info)
#
# --jaccard_clip :option, set if you have paired reads and
# you expect high gene density with UTR
# overlap (use FASTQ input file format
# for reads).
# (note: jaccard_clip is an expensive
# operation, so avoid using it unless
# necessary due to finding excessive fusion
# transcripts w/o it.)
#
# --trimmomatic :run Trimmomatic to quality trim reads
# see '--quality_trimming_params' under full usage info for tailored settings.
#
#
# --no_normalize_reads :Do *not* run in silico normalization of reads. Defaults to max. read coverage of 50.
# see '--normalize_max_read_cov' under full usage info for tailored settings.
# (note, as of Sept 21, 2016, normalization is on by default)
#
#
#
# --output <string> :name of directory for output (will be
# created if it doesn't already exist)
# default( your current working directory: "/Users/bhaas/GITHUB/trinityrnaseq/trinity_out_dir"
# note: must include 'trinity' in the name as a safety precaution! )
#
# --full_cleanup :only retain the Trinity fasta file, rename as ${output_dir}.Trinity.fasta
#
# --cite :show the Trinity literature citation
#
# --version :reports Trinity version (BLEEDING_EDGE) and exits.
#
# --show_full_usage_info :show the many many more options available for running Trinity (expert usage).
#
#
###############################################################################
#
# *Note, a typical Trinity command might be:
#
# Trinity --seqType fq --max_memory 50G --left reads_1.fq --right reads_2.fq --CPU 6
#
#
# and for Genome-guided Trinity:
#
# Trinity --genome_guided_bam rnaseq_alignments.csorted.bam --max_memory 50G
# --genome_guided_max_intron 10000 --CPU 6
#
# see: /Users/bhaas/GITHUB/trinityrnaseq/sample_data/test_Trinity_Assembly/
# for sample data and 'runMe.sh' for example Trinity execution
#
# For more details, visit: http://trinityrnaseq.github.io
#
###############################################################################
Trinity performs best with strand-specific data, in which case sense and antisense transcripts can be resolved. For protocols on strand-specific RNA-Seq, see: Borodina T, Adjaye J, Sultan M. A strand-specific library preparation protocol for RNA sequencing. Methods Enzymol. 2011;500:79-98. PubMed PMID: 21943893.
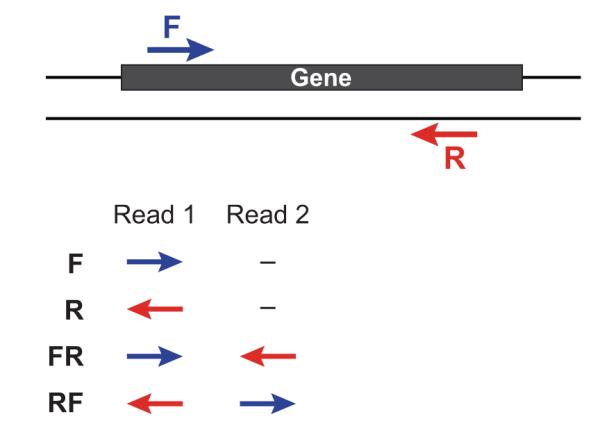
If you have strand-specific data, specify the library type. There are four library types:
- Paired reads:
- RF: first read (/1) of fragment pair is sequenced as anti-sense (reverse(R)), and second read (/2) is in the sense strand (forward(F)); typical of the dUTP/UDG sequencing method.
- FR: first read (/1) of fragment pair is sequenced as sense (forward), and second read (/2) is in the antisense strand (reverse)
- Unpaired (single) reads:
- F: the single read is in the sense (forward) orientation
- R: the single read is in the antisense (reverse) orientation
By setting the –SS_lib_type parameter to one of the above, you are indicating that the reads are strand-specific. By default, reads are treated as not strand-specific.
Other important considerations:
-
Whether you use Fastq or Fasta formatted input files, be sure to keep the reads oriented as they are reported by Illumina, if the data are strand-specific. This is because, Trinity will properly orient the sequences according to the specified library type. If the data are not strand-specific, no worries because the reads will be parsed in both orientations.
-
If you have both paired and unpaired data, and the data are NOT strand-specific, you can combine the unpaired data with the left reads of the paired fragments. Be sure that the unpaired reads have a /1 as a suffix to the accession value similarly to the left fragment reads. The right fragment reads should all have /2 as the accession suffix. Then, run Trinity using the –left and –right parameters as if all the data were paired.
-
If you have multiple paired-end library fragment sizes, set the ‘–group_pairs_distance’ according to the larger insert library. Pairings that exceed that distance will be treated as if they were unpaired by the Butterfly process.
-
by setting the ‘–CPU option’, you are indicating the maximum number of threads to be used by processes within Trinity. Note that Inchworm alone will be internally capped at 6 threads, since performance will not improve for this step beyond that setting)
Typical Trinity Command Line
A typical Trinity command for assembling non-strand-specific RNA-seq data would be like so, running the entire process on a single high-memory server (aim for ~1G RAM per ~1M ~76 base Illumina paired reads, but often much less memory is required):
Run Trinity like so:
Trinity --seqType fq --max_memory 50G \
--left reads_1.fq.gz --right reads_2.fq.gz --CPU 6
If you have multiple sets of fastq files, such as corresponding to multiple tissue types or conditions, etc., you can indicate them to Trinity like so:
Trinity --seqType fq --max_memory 50G \
--left condA_1.fq.gz,condB_1.fq.gz,condC_1.fq.gz \
--right condA_2.fq.gz,condB_2.fq.gz,condC_2.fq.gz \
--CPU 6
or better yet, create a ‘samples.txt’ file that describes the data like so:
# --samples_file <string> tab-delimited text file indicating biological replicate relationships.
# ex.
# cond_A cond_A_rep1 A_rep1_left.fq A_rep1_right.fq
# cond_A cond_A_rep2 A_rep2_left.fq A_rep2_right.fq
# cond_B cond_B_rep1 B_rep1_left.fq B_rep1_right.fq
# cond_B cond_B_rep2 B_rep2_left.fq B_rep2_right.fq
This same samples file can then be used later on with other downstream analysis steps, including expression quantification and differential expression analysis.
note that fastq files can be gzip-compressed as shown above, in which case they should require a ‘.gz’ extension.
Sample file
$ cd ../
$ wget https://www.dropbox.com/s/q1c7t4xf5kkk5um/samples.txt
| Sample name | stress | replicate | pair |
|---|---|---|---|
| KRWT | D | 1 | _1 |
Run trimming
$ trim_galore --paired --three_prime_clip_R1 20 --three_prime_clip_R2 20 --cores 2 --max_n 40 --gzip -o trimmed_fastq fastq/WTD1_1.fastq.gz fastq/WTD1_2.fastq.gz
.
.
.
.
.
$ multiqc . -n rnaseq_data --exclude rsem --exclude gatk
PLEASE CHECK YOUR MULTIQC
Trinity run
htop
cd /data/gpfs/assoc/bch709/YOURID/rnaseq/transcriptome_assembly
Trinity --seqType fq --max_memory 6G --samples_file samples.txt
Paper need to read https://academic.oup.com/gigascience/article/8/5/giz039/5488105 https://www.nature.com/articles/nbt.1883 https://www.nature.com/articles/nprot.2013.084
Assignment
- de Bruijn graph construction (10 pts)
- Draw (by hand) the de Bruijn graph for the following reads using k=3 (assume all reads are from the forward strand, no sequencing errors)
ATG
AGT
CAT
GTA
GTT
TGT
TAG
TTA
TAC- Trimming Practice
- Make
Trimfolder under/data/gpfs/assoc/bch709/YOURID/BCH709_assignment- Change directory to the folder
Trim- Copy all fastq.gz files in
/data/gpfs/assoc/bch709/Course_material/2020/RNASeq_raw_fastatoTrimfolder- Run
Trim-Galoreand processMultiQC- Generate 6 trimmed output from
MultiQCand uploadhtmlfile to WebCanvas. ** IF YOU USE “LOOP” FOR THIS JOB, YOU WILL GET ADDITIONAL 10 POINTS **
Reference:
- Conda documentation https://docs.conda.io/en/latest/
- Conda-forge https://conda-forge.github.io/
- BioConda https://bioconda.github.io/