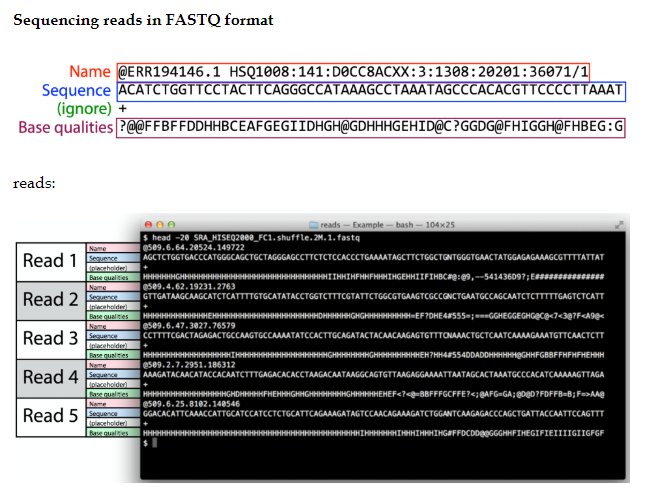
Fastq file

Connect to Pronghorn
ssh <yourID>@pronghorn.rc.unr.edu
Conda
- Dependencies is one of the main reasons to use Conda. Sometimes, install a package is not as straight forward as you think. Imagine a case like this: You want to install package Matplotlib, when installing, it asks you to install Numpy, and Scipy, because Matplotlib need these Numpy and Scipy to work. They are called the dependencies of Matplotlib. For Numpy and Scipy, they may have their own dependencies. These require even more packages.
Conda env clean
conda clean --all
Conda create enviroment
conda create -n review python=3
Conda activate enviroment
conda activate review
Example Add conda channel
Bioconda is another channel of conda, focusing on bioinformatics software. Instead of adding “-c” to search a channel only one time, “add channels” tells Conda to always search in this channel, so you don’t need to specify the channel every time. Remember to add channel in this order, so that Bioconda channel has the highest priority. Channel orders will be explained in next part.
conda config --add channels conda-forge
conda config --add channels defaults
conda config --add channels r
conda config --add channels bioconda
Install software
conda install -c bioconda trinity samtools 'multiqc<1.34' fastqc rsem jellyfish bowtie2 salmon trim-galore fastqc bioconductor-ctc bioconductor-deseq2 bioconductor-edger bioconductor-biobase bioconductor-qvalue r-ape r-gplots r-fastcluster
conda install -c anaconda openblas
conda install nano
conda install -c eumetsat tree
conda install -c lmfaber transrate
Check installation
conda list
Basic Unix/Linux command
cd
cd /data/gpfs/assoc/bch709/<YOUR_FOLDER>
mkdir
mkdir RNASEQ_REVIEW
cd RNASEQ_REVIEW
pwd
pwd
wget
file download
wget https://www.dropbox.com/s/o8r3279n5grn8el/fastq.tar https://www.dropbox.com/s/mlyrk2osnoo47em/fastq.zip
Decompress tar file
tar xvf fastq.tar
ls
Decompress zip file
unzip fastq.zip
ls
gz file
zcat
pipe \|
wc
rm
Make nano useful
nano ~/.nanorcset nowrapWhole line deletion
ctrl + kUndo
ctrl + u
Job Submission to SLURM
The SBATCH directives must appear at the top of the submission file, before any other line except for the very first line which should be the shebang (e.g. #!/bin/bash). The script itself is a job step. Other job steps are created with the srun command. For instance, the following script, hypothetically named submit.sh,
nano submit1.sh
#!/bin/bash
#SBATCH --job-name=test
#SBATCH --cpus-per-task=16
#SBATCH --time=10:00
#SBATCH --mem=1g
#SBATCH --mail-type=begin
#SBATCH --mail-type=end
#SBATCH --mail-user=<YOUR ID>@nevada.unr.edu
echo "Hello Pronghorn"
Permission change
chmod 775 submit1.sh
Check your job submission output
cat slurm<JOBID>.out
job submission 2
nano submit2.sh
#!/bin/bash
#SBATCH --job-name=test
#SBATCH --cpus-per-task=16
#SBATCH --time=10:00
#SBATCH --mem=1g
#SBATCH --mail-type=begin
#SBATCH --mail-type=fail
#SBATCH --mail-type=end
#SBATCH --mail-user=<YOUR ID>@unr.edu
#SBATCH -o hello.out # STDOUT
#SBATCH -e hello.err # STDERR
echo "Hello Pronghorn2"
Permission change
chmod 775 submit2.sh
Check your job submission output
cat hello.out
cat hello.err
Read trimming
#!/bin/bash
#SBATCH --job-name=test
#SBATCH --cpus-per-task=20
#SBATCH --time=2:00:00
#SBATCH --mem=100g
#SBATCH --mail-type=all
#SBATCH --mail-user=<YOUR ID>@unr.edu
#SBATCH -o trim.out # STDOUT
#SBATCH -e trim.err # STDERR
trim_galore --paired --three_prime_clip_R1 20 --three_prime_clip_R2 20 --cores 16 --max_n 40 --gzip -o trimmed_fastq WT1_R1.fastq.gz WT1_R2.fastq.gz
trim_galore --paired --three_prime_clip_R1 20 --three_prime_clip_R2 20 --cores 16 --max_n 40 --gzip -o trimmed_fastq WT2_R1.fastq.gz WT2_R2.fastq.gz
.
.
.
.
.
squeue
The squeue command shows the list of jobs which are currently running (they are in the RUNNING state, noted as ‘R’) or waiting for resources (noted as ‘PD’, short for PENDING).
squeue
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
983204 cpu-s2-co neb_K jzhang23 R 6-09:05:47 1 cpu-6
983660 cpu-s2-co RT3.sl yinghanc R 12:56:17 1 cpu-9
983659 cpu-s2-co RT4.sl yinghanc R 12:56:21 1 cpu-8
983068 cpu-s2-co Gd-bound dcantu R 7-06:16:01 2 cpu-[78-79]
983067 cpu-s2-co Gd-unbou dcantu R 1-17:41:56 2 cpu-[1-2]
983472 cpu-s2-co ub-all dcantu R 3-10:05:01 2 cpu-[4-5]
982604 cpu-s1-pg wrap wyim R 12-14:35:23 1 cpu-49
983585 cpu-s1-pg wrap wyim R 1-06:28:29 1 cpu-48
983628 cpu-s1-pg wrap wyim R 13:44:46 1 cpu-49
Check fastq statistics
#!/bin/bash
#SBATCH --job-name=test
#SBATCH --cpus-per-task=16
#SBATCH --time=2:00:00
#SBATCH --mem=20g
#SBATCH --mail-type=begin
#SBATCH --mail-type=end
#SBATCH --mail-user=<YOUR ID>@unr.edu
#SBATCH -o fastqc.out # STDOUT
#SBATCH -e fastqc.err # STDERR
fastqc trimmed_fastq/WT1_R1_val_1.fq.gz trimmed_fastq/WT1_R2_val_2.fq.gz .............
MultiQC
multiqc . -n rnaseq_review --exclude rsem --exclude gatk
Transfer file
multiqc . -n rnaseq_review --exclude rsem --exclude gatk
- from a remote system to local
scp username@pronghorn.rc.unr.edu:<somedirectory>/<sourcefile> <LOCAL destination>
Run Trinity
Trinity
Merge gz file previous method
zcat trimmed_fastq/WT1_R1_val_1.fq.gz ........... >> merged_R1.fastq
zcat trimmed_fastq/WT1_R2_val_2.fq.gz ........... >> merged_R2.fastq
Type sample file
nano sample.txt
###^ means CTRL key
###M- means ALT key
WT<TAB>WT_REP1<TAB>trimmed_fastq/WT1_R1_val_1.fq.gz<TAB>trimmed_fastq/WT1_R2_val_2.fq.gz
WT<TAB>WT_REP2<TAB>trimmed_fastq/WT2_R1_val_1.fq.gz<TAB>trimmed_fastq/WT2_R2_val_2.fq.gz
WT<TAB>WT_REP3<TAB>trimmed_fastq/WT3_R1_val_1.fq.gz<TAB>trimmed_fastq/WT3_R2_val_2.fq.gz
DT<TAB>DT_REP1<TAB>trimmed_fastq/DT1_R1_val_1.fq.gz<TAB>trimmed_fastq/DT1_R2_val_2.fq.gz
DT<TAB>DT_REP2<TAB>trimmed_fastq/DT2_R1_val_1.fq.gz<TAB>trimmed_fastq/DT2_R2_val_2.fq.gz
DT<TAB>DT_REP3<TAB>trimmed_fastq/DT3_R1_val_1.fq.gz<TAB>trimmed_fastq/DT3_R2_val_2.fq.gz
Trinity run previous method
nano trinity.sh
#!/bin/bash
#SBATCH --job-name=Trinity
#SBATCH --cpus-per-task=64
#SBATCH --time=2:00:00
#SBATCH --mem=100g
#SBATCH --mail-type=all
#SBATCH --mail-user=<YOUR ID>@unr.edu
#SBATCH -o trinity.out # STDOUT
#SBATCH -e trinity.err # STDERR
Trinity --seqType fq --CPU 64 --max_memory 100G --left <merged_R1.fastq> --right <merged_R2.fastq>
Trinity run current method
nano trinity.sh
#!/bin/bash
#SBATCH --job-name=Trinity
#SBATCH --cpus-per-task=64
#SBATCH --time=2:00:00
#SBATCH --mem=100g
#SBATCH --mail-type=all
#SBATCH --mail-user=<YOUR ID>@unr.edu
#SBATCH -o trinity.out # STDOUT
#SBATCH -e trinity.err # STDERR
Trinity --seqType fq --CPU 64 --max_memory 100G --samples_file sample.txt
Check your job submission output
cat trinity.out
cat trinity.err
Check folder
tree
Check your Trinity output
TrinityStats.pl
Transrate job script
#!/bin/bash
#SBATCH --job-name=Transrate
#SBATCH --cpus-per-task=32
#SBATCH --time=2:00:00
#SBATCH --mem=100g
#SBATCH --mail-type=all
#SBATCH --mail-user=<YOUR ID>@unr.edu
#SBATCH -o trinity.out # STDOUT
#SBATCH -e trinity.err # STDERR
transrate --assembly=<Trinity.fasta> --left=<merged_R1.fastq> --right=<merged_R2.fastq> --threads=32
Pre assembled result0
/data/gpfs/assoc/bch709/spiderman/RNASEQ_REVIEW/trinity_out_dir/Trinity.fasta
Run alignment
#!/bin/bash
#SBATCH --job-name=<JOB_NAME>
#SBATCH --cpus-per-task=32
#SBATCH --time=2:00:00
#SBATCH --mem=64g
#SBATCH --mail-type=begin
#SBATCH --mail-type=end
#SBATCH --mail-user=<YOUR ID>@nevada.unr.edu
#SBATCH -o <JOB_NAME>.out # STDOUT
#SBATCH -e <JOB_NAME>.err # STDERR
align_and_estimate_abundance.pl --thread_count 32 --transcripts <Trinity.fasta> --seqType fq --est_method RSEM --aln_method bowtie2 --trinity_mode --prep_reference --output_dir rsem_outdir --samples_file sample.txt
abundance_estimates_to_matrix
abundance_estimates_to_matrix.pl --est_method RSEM --gene_trans_map none --name_sample_by_basedir --cross_sample_norm TMM WT_REP1/RSEM.isoforms.results WT_REP2/RSEM.isoforms.results WT_REP3/RSEM.isoforms.results DT_REP1/RSEM.isoforms.results DT_REP2/RSEM.isoforms.results DT_REP3/RSEM.isoforms.results
PtR (Quality Check Your Samples and Biological Replicates)
Once you’ve performed transcript quantification for each of your biological replicates, it’s good to examine the data to ensure that your biological replicates are well correlated, and also to investigate relationships among your samples. If there are any obvious discrepancies among your sample and replicate relationships such as due to accidental mis-labeling of sample replicates, or strong outliers or batch effects, you’ll want to identify them before proceeding to subsequent data analyses (such as differential expression).
cut -f 1,2 sample.txt >> samples_ptr.txt
PtR --matrix RSEM.isoform.counts.matrix --samples samples_ptr.txt --CPM --log2 --min_rowSums 10 --compare_replicates
PtR --matrix RSEM.isoform.counts.matrix --samples samples_ptr.txt --CPM --log2 --min_rowSums 10 --sample_cor_matrix
PtR --matrix RSEM.isoform.counts.matrix --samples samples_ptr.txt --CPM --log2 --min_rowSums 10 --center_rows --prin_comp 3
Please transfer results to your local computer
DEG calculation
run_DE_analysis.pl --matrix RSEM.isoform.counts.matrix --samples_file samples_ptr.txt --method DESeq2
run_DE_analysis.pl --matrix RSEM.isoform.counts.matrix --samples_file samples_ptr.txt --method edgeR
cd DESeq2.XXXXX.dir
analyze_diff_expr.pl --matrix ../RSEM.isoform.TMM.EXPR.matrix -P 0.001 -C 1 --samples ../samples_ptr.txt
wc -l RSEM.isoform.counts.matrix.DT_vs_WT.DESeq2.DE_results.P0.001_C1.DE.subset
cd ../
cd edgeR.XXXXX.dir
analyze_diff_expr.pl --matrix ../RSEM.isoform.TMM.EXPR.matrix -P 0.001 -C 1 --samples ../samples_ptr.txt
wc -l RSEM.isoform.counts.matrix.DT_vs_WT.edgeR.DE_results.P0.001_C1.DE.subset
Draw Venn Diagram
conda create -n venn python=2.7
conda activate venn
conda install -c bioconda bedtools intervene r-UpSetR r-corrplot r-Cairo
cd ../
pwd
# /data/gpfs/assoc/bch709/spiderman/rnaseq/DEG2
mkdir Venn
###DESeq2
cut -f 1 ../DESeq2.91008.dir/RSEM.isoform.counts.matrix.DT_vs_WT.DESeq2.DE_results.P0.001_C1.DT-UP.subset | grep -v sample > DESeq.UP.subset
cut -f 1 ../DESeq2.91008.dir/RSEM.isoform.counts.matrix.DT_vs_WT.DESeq2.DE_results.P0.001_C1.WT-UP.subset | grep -v sample > DESeq.DOWN.subset
###edgeR
cut -f 1 ../edgeR.91693.dir/RSEM.isoform.counts.matrix.DT_vs_WT.edgeR.DE_results.P0.001_C1.DT-UP.subset | grep -v sample > edgeR.UP.subset
cut -f 1 ../edgeR.91693.dir/RSEM.isoform.counts.matrix.DT_vs_WT.edgeR.DE_results.P0.001_C1.WT-UP.subset | grep -v sample > edgeR.DOWN.subset
### Drawing
intervene venn -i DESeq.DOWN.subset DESeq.UP.subset edgeR.DOWN.subset edgeR.UP.subset --type list --save-overlaps
intervene upset -i DESeq.DOWN.subset DESeq.UP.subset edgeR.DOWN.subset edgeR.UP.subset --type list --save-overlaps
intervene pairwise -i DESeq.DOWN.subset DESeq.UP.subset edgeR.DOWN.subset edgeR.UP.subset --type list
Reference:
- Conda documentation https://docs.conda.io/en/latest/
- Conda-forge https://conda-forge.github.io/
- BioConda https://bioconda.github.io/