Chromosome Conformation Scaffolding
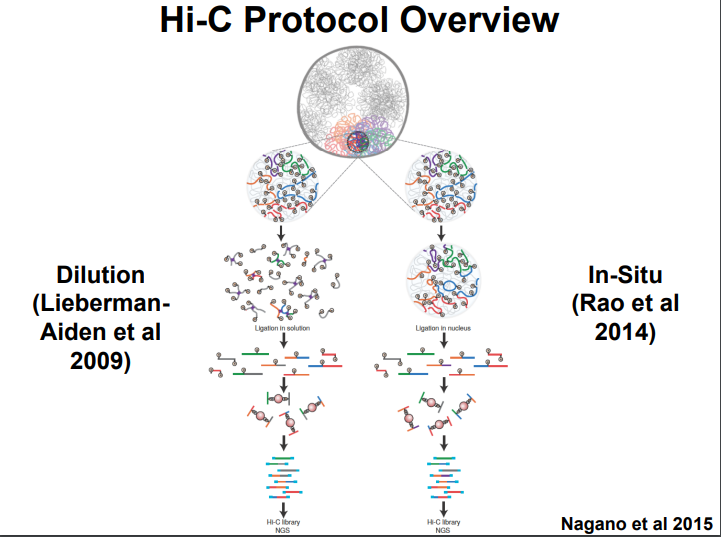
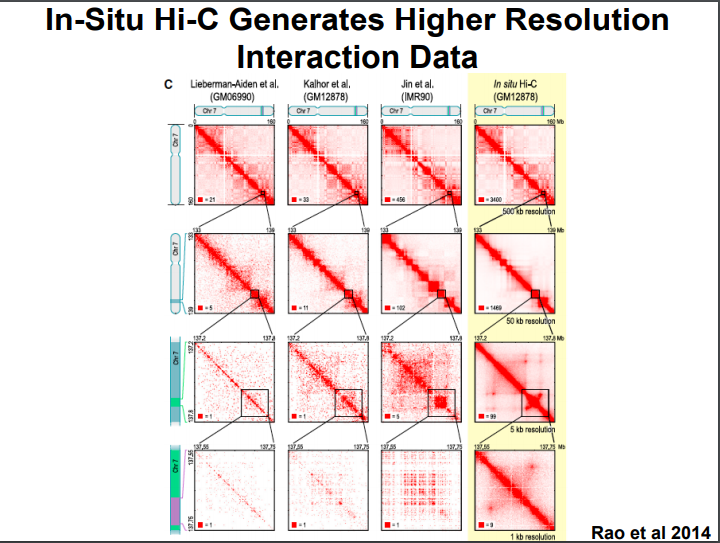
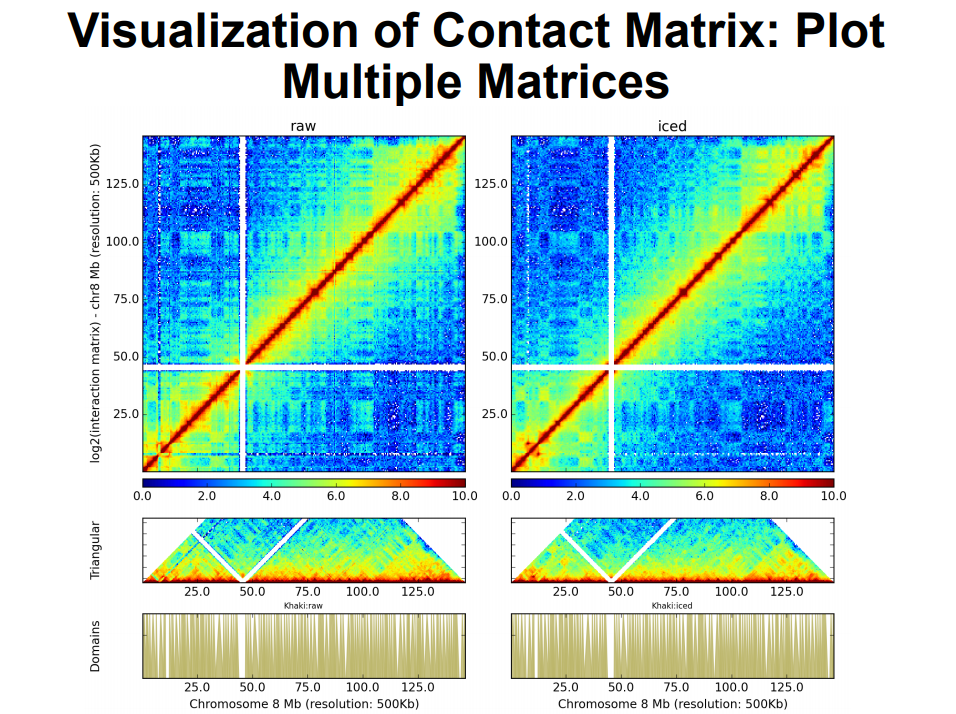
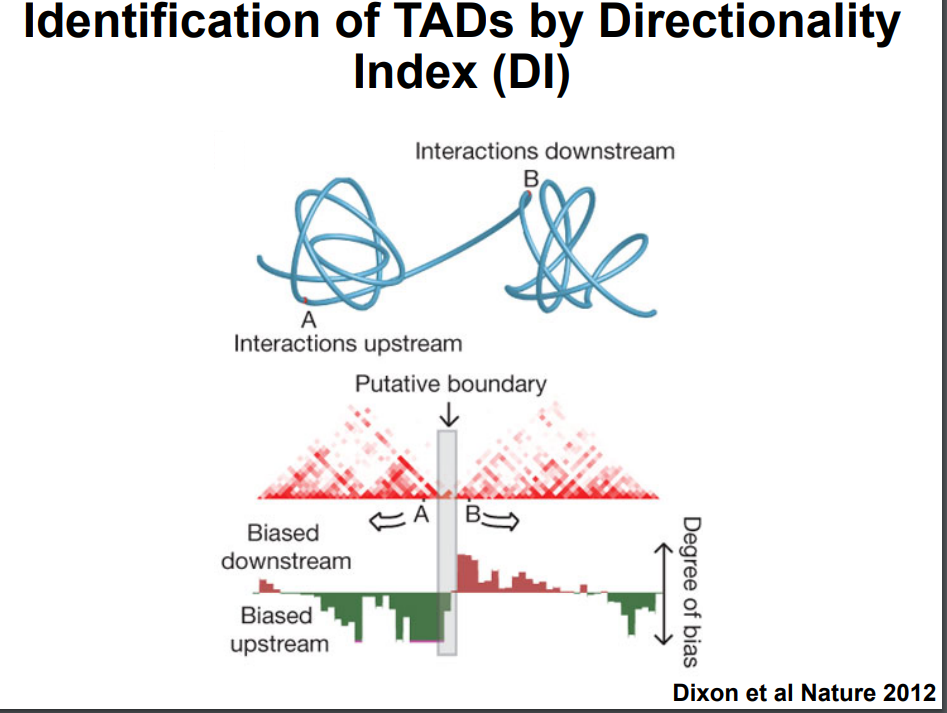
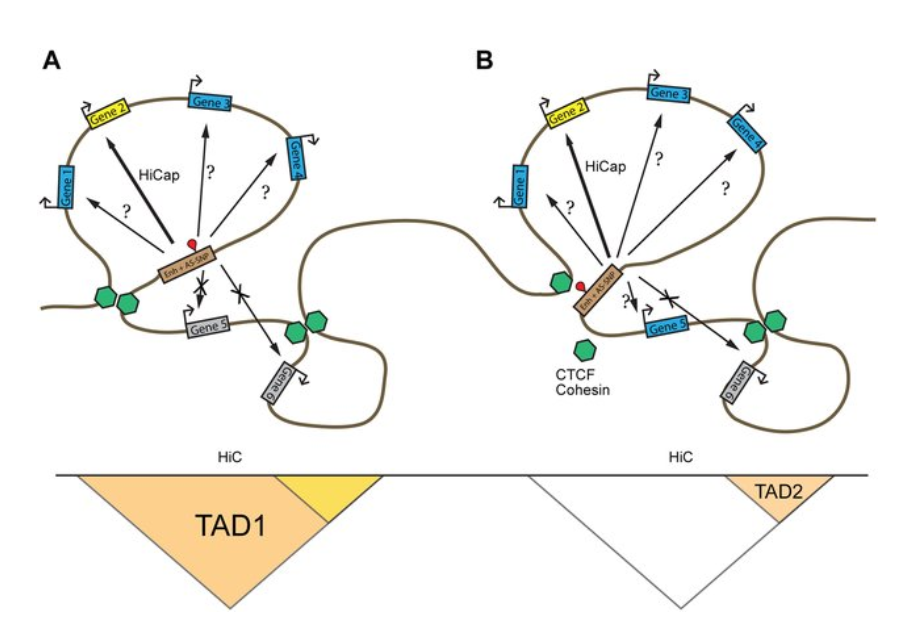
HiC for Genome Assembly
Chromosome assembly
mkdir /data/gpfs/assoc/bch709/<YOURID>/Genome_assembly/Hic
cd !$
HiC
conda create -n hic
conda activate hic
conda install -c r -c conda-forge -c anaconda -c bioconda samtools bedtools matplotlib numpy scipy bwa openssl=1.0 -y
canu.illumina.fasta
The input of HiC is the output of Pilon. If you don’t have it please do Pilon first.
File preparation
cp /data/gpfs/assoc/bch709/Course_material/2020/Genome_assembly/hic_r{1,2}.fastq.gz .
git clone https://github.com/tangerzhang/ALLHiC
cd ALLHiC
chmod 775 bin/*
chmod 775 scripts/*
cd ../
ALLHiC
Phasing and scaffolding polyploid genomes based on Hi-C data
Introduction
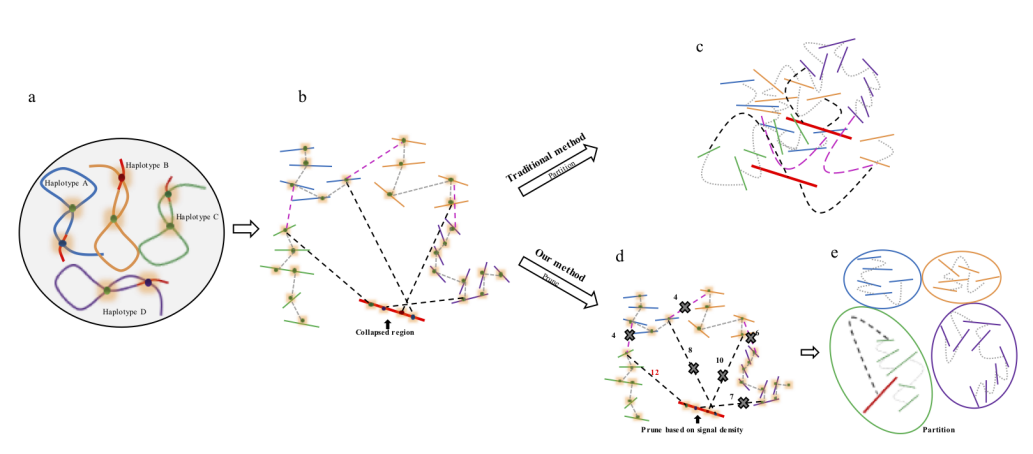
The major problem of scaffolding polyploid genome is that Hi-C signals are frequently detected between allelic haplotypes and any existing stat of art Hi-C scaffolding program links the allelic haplotypes together. To solve the problem, we developed a new Hi-C scaffolding pipeline, called ALLHIC, specifically tailored to the polyploid genomes. ALLHIC pipeline contains a total of 5 steps: prune, partition, rescue, optimize and build.
Overview of ALLHiC

Figure 1. Overview of major steps in ALLHiC algorithm. The newly released ALLHiC pipeline contains a total of 5 functions: prune, partition, rescue, optimize and build. Briefly, the prune step removes the inter-allelic links so that the homologous chromosomes are more easily separated individually. The partition function takes pruned bam file as input and clusters the linked contigs based on the linkage suggested by Hi-C, presumably along the same homologous chromosome in a preset number of partitions. The rescue function searches for contigs that are not involved in partition step from original un-pruned bam files and assigned them to specific clusters according Hi-C signal density. The optimize step takes each partition, and optimize the ordering and orientations for all the contigs. Finally, the build step reconstructs each chromosome by concatenating the contigs, adding gaps between the contigs and generating the final genome release in FASTA format.
Explanation of Prune
Prune function will firstly allow us to detect allelic contigs, which can be achieved by identifying syntenic genes based on a well-assembled close related species or an assembled monoploid genome. Signals (normalized Hi-C reads) between allelic contigs are removed from the input BAM files. In polyploid genome assembly, haplotypes that share high similarity are likely to be collapsed. Signals between the collapsed regions and nearby phased haplotypes result in chimeric scaffolds. In the prune step, only the best linkage between collapsed coting and phased contig is retained.
 Figure 2. Description of Hi-C scaffolding problem in polyploid genome and application of prune approach for haplotype phasing. (a) a schematic diagram of auto-tetraploid genome. Four homologous chromosomes are indicated as different colors (blue, orange, green and purple, respectively). Red regions in the chromosomes indicate sequences with high similarity. (b) Detection of Hi-C signals in the auto-tetraploid genome. Black dash lines indicate Hi-C signals between collapsed regions and un-collpased contigs. Pink dash lines indicate inter-haplotype Hi-C links and grey dash lines indicate intra-haplotype Hi-C links. During assembly, red regions will be collapsed due to high sequence similarity; while, other regions will be separated into different contigs if they have abundant variations. Since the collapsed regions are physically related with contigs from different haplotypes, Hi-C signals will be detected between collapsed regions with all other un-collapsed contigs. (c) Traditional Hi-C scaffolding methods will detect signals among contigs from different haplotypes as well as collapsed regions and cluster all the sequences together. (d) Prune Hi-C signals: 1- remove signals between allelic regions; 2- only retain the strongest signals between collapsed regions and un-collapsed contigs. (e) Partition based on pruned Hi-C information. Contigs are ideally phased into different groups based on prune results.
Figure 2. Description of Hi-C scaffolding problem in polyploid genome and application of prune approach for haplotype phasing. (a) a schematic diagram of auto-tetraploid genome. Four homologous chromosomes are indicated as different colors (blue, orange, green and purple, respectively). Red regions in the chromosomes indicate sequences with high similarity. (b) Detection of Hi-C signals in the auto-tetraploid genome. Black dash lines indicate Hi-C signals between collapsed regions and un-collpased contigs. Pink dash lines indicate inter-haplotype Hi-C links and grey dash lines indicate intra-haplotype Hi-C links. During assembly, red regions will be collapsed due to high sequence similarity; while, other regions will be separated into different contigs if they have abundant variations. Since the collapsed regions are physically related with contigs from different haplotypes, Hi-C signals will be detected between collapsed regions with all other un-collapsed contigs. (c) Traditional Hi-C scaffolding methods will detect signals among contigs from different haplotypes as well as collapsed regions and cluster all the sequences together. (d) Prune Hi-C signals: 1- remove signals between allelic regions; 2- only retain the strongest signals between collapsed regions and un-collapsed contigs. (e) Partition based on pruned Hi-C information. Contigs are ideally phased into different groups based on prune results.
Citations
Zhang, X. Zhang, S. Zhao, Q. Ming, R. Tang, H. Assembly of allele-aware, chromosomal scale autopolyploid genomes based on Hi-C data. Nature Plants, doi:10.1038/s41477-019-0487-8 (2019).
Zhang, J. Zhang, X. Tang, H. Zhang, Q. et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nature Genetics, doi:10.1038/s41588-018-0237-2 (2018).
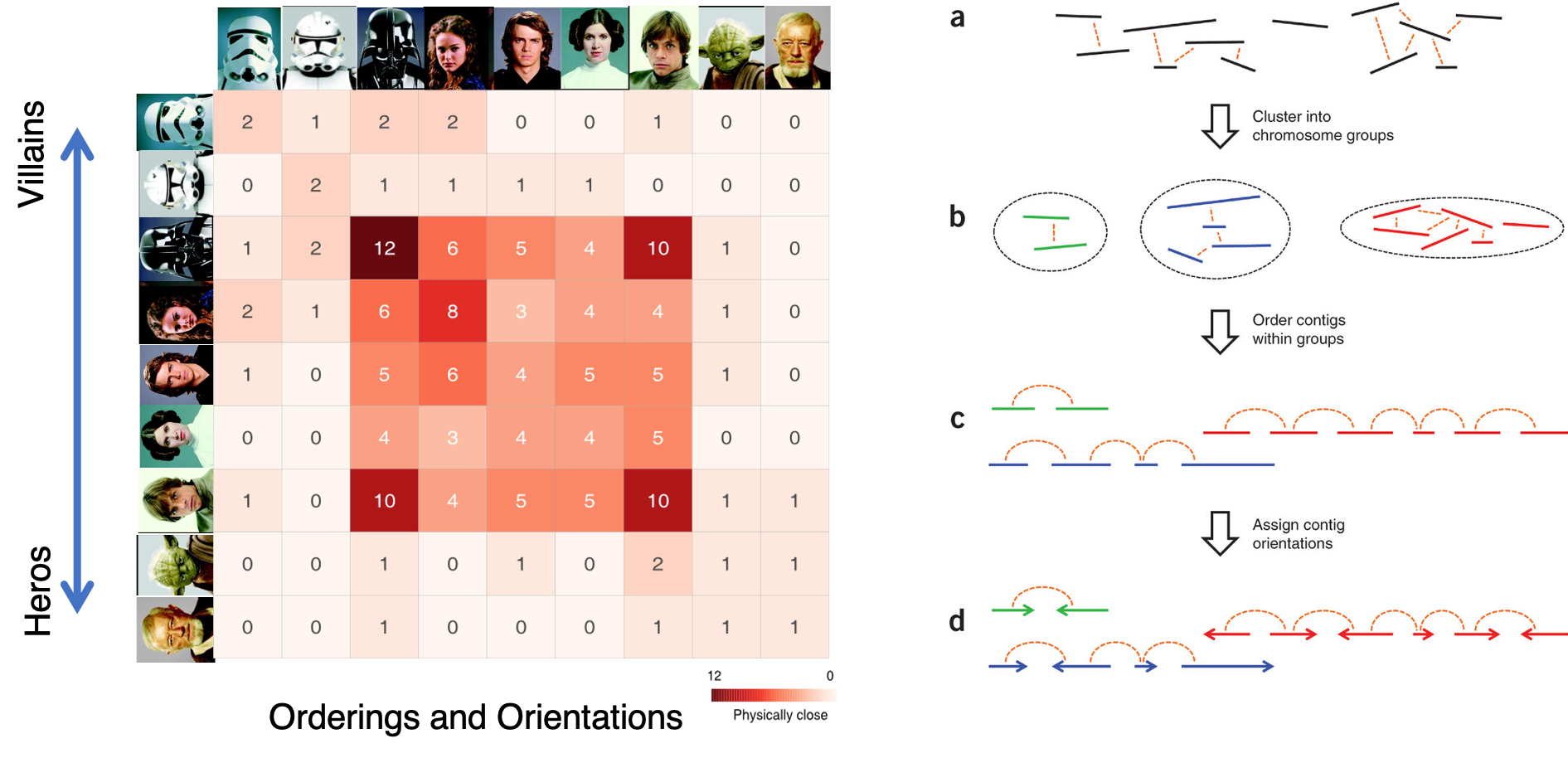
Algorithm demo
Solving scaffold ordering and orientation (OO) in general is NP-hard. ALLMAPS converts the problem into Traveling Salesman Problem (TSP) and refines scaffold OO using Genetic Algorithm. For rough idea, a ‘live’ demo of the scaffold OO on yellow catfish chromosome 1 can be viewed in the animation below.

Traveling Salesman Problem
Run AllHIC -> hic.sh
#!/bin/bash
#SBATCH --job-name=AllHIC
#SBATCH --cpus-per-task=1
#SBATCH --time=12:00:00
#SBATCH --mem=3500M
#SBATCH --mail-type=all
#SBATCH --mail-user=<YOUREMAIL>
#SBATCH -o hic.out # STDOUT
#SBATCH -e hic.err # STDERR
#SBATCH -p cpu-s2-core-0
#SBATCH -A cpu-s2-bch709-0
export PATH=/data/gpfs/assoc/bch709/<YOURID>/Genome_assembly/Hic/ALLHiC/scripts/:/data/gpfs/assoc/bch709/<YOURID>/Genome_assembly/Hic/ALLHiC/bin/:$PATH
cp /data/gpfs/assoc/bch709/<YOURID>/Genome_assembly/Pilon/canu.illumina.fasta .
bwa index canu.illumina.fasta
bwa aln -t 1 canu.illumina.fasta hic_r1.fastq.gz > sample_R1.sai
bwa aln -t 1 canu.illumina.fasta hic_r2.fastq.gz > sample_R2.sai
bwa sampe canu.illumina.fasta sample_R1.sai sample_R2.sai hic_r1.fastq.gz hic_r2.fastq.gz > sample.bwa_aln.sam
PreprocessSAMs.pl sample.bwa_aln.sam canu.illumina.fasta MBOI
filterBAM_forHiC.pl sample.bwa_aln.REduced.paired_only.bam sample.clean.sam
samtools view -bht canu.illumina.fasta.fai sample.bwa_aln.sam > sample.clean.bam
cp sample.clean.bam hic.bam
allhic extract --minLinks 10 hic.bam canu.illumina.fasta
allhic partition hic.counts_GATC.txt hic.pairs.txt 2
allhic optimize hic.counts_GATC.2g1.txt hic.clm
allhic optimize hic.counts_GATC.2g2.txt hic.clm
ALLHiC_build canu.illumina.fasta
cp groups.asm.fasta bch709_assembly.fasta
samtools faidx bch709_assembly.fasta
cut -f 1,2 bch709_assembly.fasta.fai | egrep hic > chrn.list
ALLHiC_plot hic.bam groups.agp chrn.list 10k pdf
](/fig/hicmovie.gif)
Allhic result
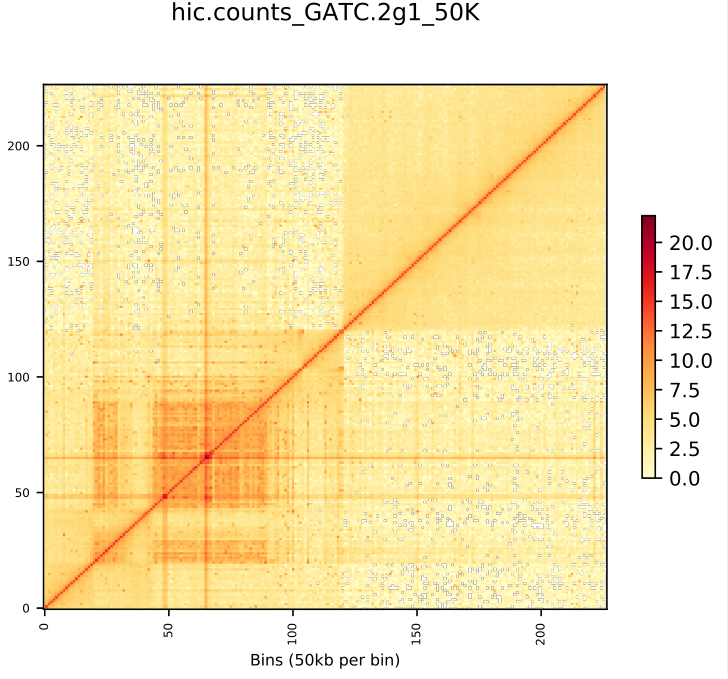
Original Sequence
https://www.dropbox.com/s/xpnhj6j99dr1kum/Athaliana_subset_BCH709.fa
set the environment
conda activate genomeassembly
Run Genome Wide Global Alignmnet Software
#!/bin/bash
#SBATCH --job-name=nucmer
#SBATCH --cpus-per-task=2
#SBATCH --time=15:00
#SBATCH --mem=10g
#SBATCH --mail-type=all
#SBATCH --mail-user=<YOURID>@unr.edu
#SBATCH -o nucmer.out # STDOUT
#SBATCH -e nucmer.err # STDERR
#SBATCH --account=cpu-s6-test-0
#SBATCH --partition=cpu-s6-test-0
nucmer --coords -p bch709_assembly_vs_original_sequence bch709_assembly.fasta Athaliana_subset_BCH709.fa
wget https://dnanexus.github.io/dot/DotPrep.py
chmod 775 DotPrep.py
nano DotPrep.sh
#!/bin/bash
#SBATCH --job-name=dot
#SBATCH --cpus-per-task=2
#SBATCH --time=15:00
#SBATCH --mem=10g
#SBATCH --mail-type=all
#SBATCH --mail-user=wyim@unr.edu
#SBATCH -o nucmer.out # STDOUT
#SBATCH -e nucmer.err # STDERR
#SBATCH --account=cpu-s6-test-0
#SBATCH --partition=cpu-s6-test-0
python DotPrep.py --delta bch709_assembly_vs_original_sequence.delta --out bch709_assembly_vs_original_sequence
DOT plot
https://dnanexus.github.io/dot/
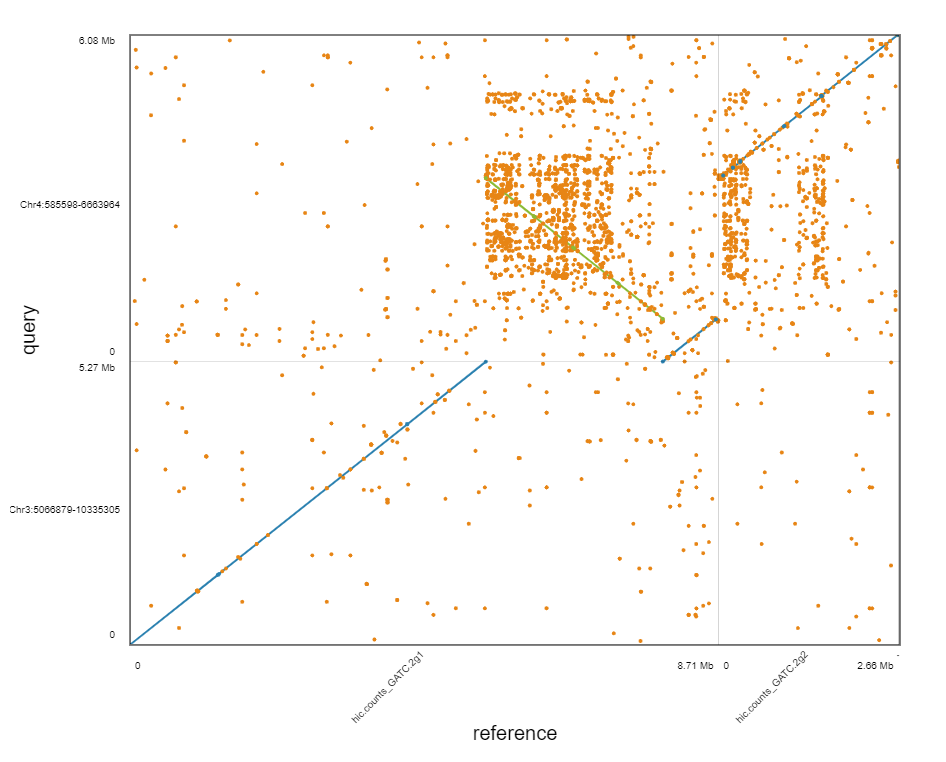
What is the problem?