HPC Clusters
This exercise focuses on using HPC clusters for large-scale data analysis (e.g., Next Generation Sequencing, genome annotation, evolutionary studies). These clusters contain multiple processors with large amounts of RAM, making them ideal for computationally intensive tasks. The operating system is primarily UNIX, accessed via the command line. All the commands you’ve learned in previous exercises can be used here.
Pronghorn High Performance Computing offers shared infrastructure for researchers and students at UNR. You can find available resources here. Request access through your department or advisor. All attendees of this workshop will have their accounts set up on the HPC class education cluster using their UNR NetID and password. You should have received a confirmation email with connection instructions. This exercise covers connecting to a remote HPC server, transferring files, and running programs by requesting resources.
To log into the HPC front-end/job-submission system (pronghorn.rc.unr.edu), use your UNR NetID and password. Windows users will need an SSH client, while Mac/Linux users have SSH built-in.
ssh <YOUR_NET_ID>@pronghorn.rc.unr.edu
## First login will prompt for key confirmation. Choose 'yes.'
Pronghorn HPC Cluster Overview
Pronghorn is UNR’s new GPU-accelerated HPC cluster, supporting general research across NSHE. Comprising CPU, GPU, and storage subsystems, Pronghorn’s main features include:
- CPU Partition: 93 nodes, 2,976 CPU cores, 21TiB of memory.
- GPU Partition: 44 NVIDIA Tesla P100 GPUs, 352 CPU cores, 2.75TiB of memory.
- Storage: 1PB high-performance storage using IBM SpectrumScale.
Pronghorn is located at Switch Citadel Campus, 25 miles east of UNR. Switch is renowned for sustainable data center operations.
Customizing the Bash Prompt for Pronghorn
echo '###BCH709' >> ~/.bashrc
echo 'tty -s && export PS1="\[\033[38;5;164m\]\u\[$(tput sgr0)\]\[\033[38;5;15m\] \[$(tput sgr0)\]\[\033[38;5;231m\]@\[$(tput sgr0)\]\[\033[38;5;15m\] \[$(tput sgr0)\]\[\033[38;5;2m\]\h\[$(tput sgr0)\]\[\033[38;5;15m\] \[$(tput sgr0)\]\[\033[38;5;172m\]\t\[$(tput sgr0)\]\[\033[38;5;15m\] \[$(tput sgr0)\]\[\033[38;5;2m\]\w\[$(tput sgr0)\]\[\033[38;5;15m\]\n \[$(tput sgr0)\]"' >> ~/.bashrc
echo "alias ls='ls --color=auto'" >> ~/.bashrc
source ~/.bashrc
File Transfer Methods
Several methods exist for transferring files to/from HPC clusters, including command-line tools (scp, rsync) and graphical clients (SCP, SFTP). For secure copying:
Transferring from Local to Remote System
scp <source_file> <username>@pronghorn.rc.unr.edu:<target_location>
Example:
mkdir ~/bch709
cd ~/bch709
echo "hello world" > test_uploading_file.txt
scp test_uploading_file.txt <username>@pronghorn.rc.unr.edu:~/
Transferring from Remote to Local System
scp <username>@pronghorn.rc.unr.edu:<source_file> <destination_file>
Example:
scp <username>@pronghorn.rc.unr.edu:~/test_downloading_file.txt ~/
Recursive Directory Transfer (Local to Remote)
scp -r <source_directory> <username>@pronghorn.rc.unr.edu:<target_directory>
Example:
scp -r ../bch709 <username>@pronghorn.rc.unr.edu:~/
Opening Location
- Windows (WSL)
cd ~/bch709 explorer.exe . - Mac
cd ~/bch709 open .
Rsync Usage
- Sync local directory to remote server:
rsync -avhP <source_directory> <username>@pronghorn.rc.unr.edu:<target_directory> - Sync remote directory to local:
rsync -avhP <username>@pronghorn.rc.unr.edu:<source_directory> <target_directory>
Conda Installation on Pronghorn
Install Miniconda3 using the following commands:
curl -O https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
bash Miniconda3-latest-Linux-x86_64.sh
~/miniconda3/bin/conda init
Follow the on-screen instructions to complete the installation.
Using Conda Environment
conda install mamba
mamba create -y -n RNASEQ_bch709 -c bioconda -c conda-forge sra-tools=3.1.1 minimap2 star trim-galore gffread seqkit samtools 'multiqc<1.34' subread tree
conda activate RNASEQ_bch709
Environment create and installation in Pronghorn
# Create a new conda environment named "rnaseq".
# Add two channels to fetch the required packages:
# - bioconda: A channel specializing in bioinformatics software
# - conda-forge: A community-maintained collection of conda packages
-c bioconda -c conda-forge
# List of packages/software to be installed in the "rnaseq" environment:
# - fastqc: A tool for quality control checks on raw sequence data
# - trim-galore: A wrapper tool around Cutadapt and FastQC to consistently apply adapter and quality trimming
# - hisat2: A fast and sensitive alignment program for mapping next-generation sequencing reads to a population of genomes
# - samtools: A suite of programs for interacting with high-throughput sequencing data
# - subread: A toolkit for processing next-gen sequencing read data, including feature counting
# - bioconductor-deseq2: A package for differential expression analysis based on the negative binomial distribution
# - bc: An arbitrary precision calculator language
# The "-y" flag allows the command to proceed without asking for user confirmation.
## Connecting Scratch Disk
```bash
mkdir /data/gpfs/assoc/bch709-5/students/$(whoami)
cd /data/gpfs/assoc/bch709-5/students/$(whoami)
ln -s /data/gpfs/assoc/bch709-5/students/$(whoami) ~/scratch
Job Submission with SBATCH
Create a job submission script named submit.sh:
nano submit.sh
Add the following content:
#!/bin/bash
#SBATCH --job-name=test
#SBATCH --mail-type=ALL
#SBATCH --mail-user=<YOUR_EMAIL>
#SBATCH --ntasks=1
#SBATCH --mem=1g
#SBATCH --time=8:10:00
#SBATCH --account=cpu-s5-bch709-5
#SBATCH --partition=cpu-core-0
for i in {1..1000}; do
echo $i;
sleep 1;
done
Submit the job:
chmod 775 submit.sh
sbatch submit.sh
To cancel the job:
scancel <JOB_ID>
SRA
Sequence Read Archive (SRA) data, available through multiple cloud providers and NCBI servers, is the largest publicly available repository of high throughput sequencing data. The archive accepts data from all branches of life as well as metagenomic and environmental surveys.
Searching the SRA: Searching the SRA can be complicated. Often a paper or reference will specify the accession number(s) connected to a dataset. You can search flexibly using a number of terms (such as the organism name) or the filters (e.g. DNA vs. RNA). The SRA Help Manual provides several useful explanations. It is important to know is that projects are organized and related at several levels, and some important terms include:
Bioproject: A BioProject is a collection of biological data related to a single initiative, originating from a single organization or from a consortium of coordinating organizations; see for example Bio Project 272719 Bio Sample: A description of the source materials for a project Run: These are the actual sequencing runs (usually starting with SRR); see for example SRR1761506
Publication (Arabidopsis)
SRA Bioproject site
https://www.ncbi.nlm.nih.gov/bioproject/PRJNA272719
Runinfo
| Run | ReleaseDate | LoadDate | spots | bases | spots_with_mates | avgLength | size_MB | AssemblyName | download_path | Experiment | LibraryName | LibraryStrategy | LibrarySelection | LibrarySource | LibraryLayout | InsertSize | InsertDev | Platform | Model | SRAStudy | BioProject | Study_Pubmed_id | ProjectID | Sample | BioSample | SampleType | TaxID | ScientificName | SampleName | g1k_pop_code | source | g1k_analysis_group | Subject_ID | Sex | Disease | Tumor | Affection_Status | Analyte_Type | Histological_Type | Body_Site | CenterName | Submission | dbgap_study_accession | Consent | RunHash | ReadHash |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SRR1761506 | 1/15/2016 15:51 | 1/15/2015 12:43 | 7379945 | 1490748890 | 7379945 | 202 | 899 | https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/SRR1761506/SRR1761506.1 | SRX844600 | RNA-Seq | cDNA | TRANSCRIPTOMIC | PAIRED | 0 | 0 | ILLUMINA | Illumina HiSeq 2500 | SRP052302 | PRJNA272719 | 3 | 272719 | SRS820503 | SAMN03285048 | simple | 3702 | Arabidopsis thaliana | GSM1585887 | no | GEO | SRA232612 | public | F335FB96DDD730AC6D3AE4F6683BF234 | 12818EB5275BCB7BCB815E147BFD0619 | |||||||||||||
| SRR1761507 | 1/15/2016 15:51 | 1/15/2015 12:43 | 9182965 | 1854958930 | 9182965 | 202 | 1123 | https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/SRR1761507/SRR1761507.1 | SRX844601 | RNA-Seq | cDNA | TRANSCRIPTOMIC | PAIRED | 0 | 0 | ILLUMINA | Illumina HiSeq 2500 | SRP052302 | PRJNA272719 | 3 | 272719 | SRS820504 | SAMN03285045 | simple | 3702 | Arabidopsis thaliana | GSM1585888 | no | GEO | SRA232612 | public | 00FD62759BF7BBAEF123BF5960B2A616 | A61DCD3B96AB0796AB5E969F24F81B76 | |||||||||||||
| SRR1761508 | 1/15/2016 15:51 | 1/15/2015 12:47 | 19060611 | 3850243422 | 19060611 | 202 | 2324 | https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/SRR1761508/SRR1761508.1 | SRX844602 | RNA-Seq | cDNA | TRANSCRIPTOMIC | PAIRED | 0 | 0 | ILLUMINA | Illumina HiSeq 2500 | SRP052302 | PRJNA272719 | 3 | 272719 | SRS820505 | SAMN03285046 | simple | 3702 | Arabidopsis thaliana | GSM1585889 | no | GEO | SRA232612 | public | B75A3E64E88B1900102264522D2281CB | 657987ABC8043768E99BD82947608CAC | |||||||||||||
| SRR1761509 | 1/15/2016 15:51 | 1/15/2015 12:51 | 16555739 | 3344259278 | 16555739 | 202 | 2016 | https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/SRR1761509/SRR1761509.1 | SRX844603 | RNA-Seq | cDNA | TRANSCRIPTOMIC | PAIRED | 0 | 0 | ILLUMINA | Illumina HiSeq 2500 | SRP052302 | PRJNA272719 | 3 | 272719 | SRS820506 | SAMN03285049 | simple | 3702 | Arabidopsis thaliana | GSM1585890 | no | GEO | SRA232612 | public | 27CA2B82B69EEF56EAF53D3F464EEB7B | 2B56CA09F3655F4BBB412FD2EE8D956C | |||||||||||||
| SRR1761510 | 1/15/2016 15:51 | 1/15/2015 12:46 | 12700942 | 2565590284 | 12700942 | 202 | 1552 | https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/SRR1761510/SRR1761510.1 | SRX844604 | RNA-Seq | cDNA | TRANSCRIPTOMIC | PAIRED | 0 | 0 | ILLUMINA | Illumina HiSeq 2500 | SRP052302 | PRJNA272719 | 3 | 272719 | SRS820508 | SAMN03285050 | simple | 3702 | Arabidopsis thaliana | GSM1585891 | no | GEO | SRA232612 | public | D3901795C7ED74B8850480132F4688DA | 476A9484DCFCF9FFFDAADAAF4CE5D0EA | |||||||||||||
| SRR1761511 | 1/15/2016 15:51 | 1/15/2015 12:44 | 13353992 | 2697506384 | 13353992 | 202 | 1639 | https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/SRR1761511/SRR1761511.1 | SRX844605 | RNA-Seq | cDNA | TRANSCRIPTOMIC | PAIRED | 0 | 0 | ILLUMINA | Illumina HiSeq 2500 | SRP052302 | PRJNA272719 | 3 | 272719 | SRS820507 | SAMN03285047 | simple | 3702 | Arabidopsis thaliana | GSM1585892 | no | GEO | SRA232612 | public | 5078379601081319FCBF67C7465C404A | E3B4195AFEA115ACDA6DEF6E4AA7D8DF | |||||||||||||
| SRR1761512 | 1/15/2016 15:51 | 1/15/2015 12:44 | 8134575 | 1643184150 | 8134575 | 202 | 1067 | https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/SRR1761512/SRR1761512.1 | SRX844606 | RNA-Seq | cDNA | TRANSCRIPTOMIC | PAIRED | 0 | 0 | ILLUMINA | Illumina HiSeq 2500 | SRP052302 | PRJNA272719 | 3 | 272719 | SRS820509 | SAMN03285051 | simple | 3702 | Arabidopsis thaliana | GSM1585893 | no | GEO | SRA232612 | public | DDB8F763B71B1E29CC9C1F4C53D88D07 | 8F31604D3A4120A50B2E49329A786FA6 | |||||||||||||
| SRR1761513 | 1/15/2016 15:51 | 1/15/2015 12:43 | 7333641 | 1481395482 | 7333641 | 202 | 960 | https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/SRR1761513/SRR1761513.1 | SRX844607 | RNA-Seq | cDNA | TRANSCRIPTOMIC | PAIRED | 0 | 0 | ILLUMINA | Illumina HiSeq 2500 | SRP052302 | PRJNA272719 | 3 | 272719 | SRS820510 | SAMN03285053 | simple | 3702 | Arabidopsis thaliana | GSM1585894 | no | GEO | SRA232612 | public | 4068AE245EB0A81DFF02889D35864AF2 | 8E05C4BC316FBDFEBAA3099C54E7517B | |||||||||||||
| SRR1761514 | 1/15/2016 15:51 | 1/15/2015 12:44 | 6160111 | 1244342422 | 6160111 | 202 | 807 | https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/SRR1761514/SRR1761514.1 | SRX844608 | RNA-Seq | cDNA | TRANSCRIPTOMIC | PAIRED | 0 | 0 | ILLUMINA | Illumina HiSeq 2500 | SRP052302 | PRJNA272719 | 3 | 272719 | SRS820511 | SAMN03285059 | simple | 3702 | Arabidopsis thaliana | GSM1585895 | no | GEO | SRA232612 | public | 0A1F3E9192E7F9F4B3758B1CE514D264 | 81BFDB94C797624B34AFFEB554CE4D98 | |||||||||||||
| SRR1761515 | 1/15/2016 15:51 | 1/15/2015 12:44 | 7988876 | 1613752952 | 7988876 | 202 | 1048 | https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/SRR1761515/SRR1761515.1 | SRX844609 | RNA-Seq | cDNA | TRANSCRIPTOMIC | PAIRED | 0 | 0 | ILLUMINA | Illumina HiSeq 2500 | SRP052302 | PRJNA272719 | 3 | 272719 | SRS820512 | SAMN03285054 | simple | 3702 | Arabidopsis thaliana | GSM1585896 | no | GEO | SRA232612 | public | 39B37A0BD484C736616C5B0A45194525 | 85B031D74DF90AD1815AA1BBBF1F12BD | |||||||||||||
| SRR1761516 | 1/15/2016 15:51 | 1/15/2015 12:44 | 8770090 | 1771558180 | 8770090 | 202 | 1152 | https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/SRR1761516/SRR1761516.1 | SRX844610 | RNA-Seq | cDNA | TRANSCRIPTOMIC | PAIRED | 0 | 0 | ILLUMINA | Illumina HiSeq 2500 | SRP052302 | PRJNA272719 | 3 | 272719 | SRS820514 | SAMN03285055 | simple | 3702 | Arabidopsis thaliana | GSM1585897 | no | GEO | SRA232612 | public | E4728DFBF0F9F04B89A5B041FA570EB3 | B96545CB9C4C3EE1C9F1E8B3D4CE9D24 | |||||||||||||
| SRR1761517 | 1/15/2016 15:51 | 1/15/2015 12:44 | 8229157 | 1662289714 | 8229157 | 202 | 1075 | https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/SRR1761517/SRR1761517.1 | SRX844611 | RNA-Seq | cDNA | TRANSCRIPTOMIC | PAIRED | 0 | 0 | ILLUMINA | Illumina HiSeq 2500 | SRP052302 | PRJNA272719 | 3 | 272719 | SRS820513 | SAMN03285058 | simple | 3702 | Arabidopsis thaliana | GSM1585898 | no | GEO | SRA232612 | public | C05BC519960B075038834458514473EB | 4EF7877FC59FF5214DBF2E2FE36D67C5 | |||||||||||||
| SRR1761518 | 1/15/2016 15:51 | 1/15/2015 12:44 | 8760931 | 1769708062 | 8760931 | 202 | 1072 | https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/SRR1761518/SRR1761518.1 | SRX844612 | RNA-Seq | cDNA | TRANSCRIPTOMIC | PAIRED | 0 | 0 | ILLUMINA | Illumina HiSeq 2500 | SRP052302 | PRJNA272719 | 3 | 272719 | SRS820515 | SAMN03285052 | simple | 3702 | Arabidopsis thaliana | GSM1585899 | no | GEO | SRA232612 | public | 7D8333182062545CECD5308A222FF506 | 382F586C4BF74E474D8F9282E36BE4EC | |||||||||||||
| SRR1761519 | 1/15/2016 15:51 | 1/15/2015 12:44 | 6643107 | 1341907614 | 6643107 | 202 | 811 | https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/SRR1761519/SRR1761519.1 | SRX844613 | RNA-Seq | cDNA | TRANSCRIPTOMIC | PAIRED | 0 | 0 | ILLUMINA | Illumina HiSeq 2500 | SRP052302 | PRJNA272719 | 3 | 272719 | SRS820516 | SAMN03285056 | simple | 3702 | Arabidopsis thaliana | GSM1585900 | no | GEO | SRA232612 | public | 163BD8073D7E128D8AD1B253A722DD08 | DFBCC891EB5FA97490E32935E54C9E14 | |||||||||||||
| SRR1761520 | 1/15/2016 15:51 | 1/15/2015 12:44 | 8506472 | 1718307344 | 8506472 | 202 | 1040 | https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/SRR1761520/SRR1761520.1 | SRX844614 | RNA-Seq | cDNA | TRANSCRIPTOMIC | PAIRED | 0 | 0 | ILLUMINA | Illumina HiSeq 2500 | SRP052302 | PRJNA272719 | 3 | 272719 | SRS820517 | SAMN03285062 | simple | 3702 | Arabidopsis thaliana | GSM1585901 | no | GEO | SRA232612 | public | 791BD0D8840AA5F1D74E396668638DA1 | AF4694425D34F84095F6CFD6F4A09936 | |||||||||||||
| SRR1761521 | 1/15/2016 15:51 | 1/15/2015 12:46 | 13166085 | 2659549170 | 13166085 | 202 | 1609 | https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/SRR1761521/SRR1761521.1 | SRX844615 | RNA-Seq | cDNA | TRANSCRIPTOMIC | PAIRED | 0 | 0 | ILLUMINA | Illumina HiSeq 2500 | SRP052302 | PRJNA272719 | 3 | 272719 | SRS820518 | SAMN03285057 | simple | 3702 | Arabidopsis thaliana | GSM1585902 | no | GEO | SRA232612 | public | 47C40480E9B7DB62B4BEE0F2193D16B3 | 1443C58A943C07D3275AB12DC31644A9 | |||||||||||||
| SRR1761522 | 1/15/2016 15:51 | 1/15/2015 12:49 | 9496483 | 1918289566 | 9496483 | 202 | 1162 | https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/SRR1761522/SRR1761522.1 | SRX844616 | RNA-Seq | cDNA | TRANSCRIPTOMIC | PAIRED | 0 | 0 | ILLUMINA | Illumina HiSeq 2500 | SRP052302 | PRJNA272719 | 3 | 272719 | SRS820519 | SAMN03285061 | simple | 3702 | Arabidopsis thaliana | GSM1585903 | no | GEO | SRA232612 | public | BB05DF11E1F95427530D69DB5E0FA667 | 7706862FB2DF957E4041D2064A691CF6 | |||||||||||||
| SRR1761523 | 1/15/2016 15:51 | 1/15/2015 12:46 | 14999315 | 3029861630 | 14999315 | 202 | 1832 | https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/SRR1761523/SRR1761523.1 | SRX844617 | RNA-Seq | cDNA | TRANSCRIPTOMIC | PAIRED | 0 | 0 | ILLUMINA | Illumina HiSeq 2500 | SRP052302 | PRJNA272719 | 3 | 272719 | SRS820520 | SAMN03285060 | simple | 3702 | Arabidopsis thaliana | GSM1585904 | no | GEO | SRA232612 | public | 101D3A151E632224C09A702BD2F59CF5 | 0AC99FAA6B8941F89FFCBB8B1910696E |
Subset of data
| Sample information | Run |
|---|---|
| WT_rep1 | SRR1761506 |
| WT_rep2 | SRR1761507 |
| WT_rep3 | SRR1761508 |
| ABA_rep1 | SRR1761509 |
| ABA_rep2 | SRR1761510 |
| ABA_rep3 | SRR1761511 |
SRA Data Access
SRA (Sequence Read Archive) is a repository of high-throughput sequencing data. To download sequencing data from SRA, use fastq-dump.
preparing Fastq-Dump Job
Create and submit a script fastq-dump.sh to download RNA-Seq data:
mkdir ~/scratch/raw_data
cd ~/scratch/raw_data
nano fastq-dump.sh
#!/bin/bash
#SBATCH --job-name=fastqdump_ATH
#SBATCH --cpus-per-task=2
#SBATCH --time=2-15:00:00
#SBATCH --mem=16g
#SBATCH --mail-type=ALL
#SBATCH --mail-user=<YOUR_EMAIL>
#SBATCH -o fastq-dump.out
#SBATCH --account=cpu-s5-bch709-5
#SBATCH --partition=cpu-core-0
fastq-dump SRR1761506 --split-3 --outdir /data/gpfs/assoc/bch709-5/students/$(whoami)/raw_data --gzip
fastq-dump SRR1761507 --split-3 --outdir /data/gpfs/assoc/bch709-5/students/$(whoami)/raw_data --gzip
fastq-dump SRR1761508 --split-3 --outdir /data/gpfs/assoc/bch709-5/students/$(whoami)/raw_data --gzip
fastq-dump SRR1761509 --split-3 --outdir /data/gpfs/assoc/bch709-5/students/$(whoami)/raw_data --gzip
fastq-dump SRR1761510 --split-3 --outdir /data/gpfs/assoc/bch709-5/students/$(whoami)/raw_data --gzip
fastq-dump SRR1761511 --split-3 --outdir /data/gpfs/assoc/bch709-5/students/$(whoami)/raw_data --gzip
submit Fastq-Dump Job
sbatch fastq-dump.sh
squeue
Explain the command
This command uses the `fastq-dump` tool from the SRA (Sequence Read Archive) Toolkit to download and process sequencing data associated with the accession number **SRR1761510**. Let’s break it down:
1. **`fastq-dump SRR1761510`**: This part tells `fastq-dump` to retrieve the sequencing data associated with the accession **SRR1761510**. Accession numbers like these correspond to specific datasets available in the NCBI SRA database.
2. **`--split-3`**: This option splits paired-end reads into separate files (for example, `_1` for the first read in a pair and `_2` for the second read). If there are also unpaired reads, they’ll be stored in a separate file as well.
3. **`--outdir /data/gpfs/assoc/bch709-5/students/$(whoami)/raw_data`**: The `--outdir` option specifies the output directory for the downloaded FASTQ files. The path:
/data/gpfs/assoc/bch709-5/students/$(whoami)/raw_data
includes `$(whoami)`, which dynamically inserts the current username. This makes sure each user has their own dedicated output directory under `raw_data`.
4. **`--gzip`**: This option compresses the output FASTQ files in **GZIP** format, saving storage space and making downstream data handling more efficient.
In summary, this command will download the sequencing data for **SRR1761510** from SRA, split the paired-end reads, and save them in a specific directory with GZIP compression.
Trimming Reads with Trim-Galore
Submit a trimming job:
mkdir -p ~/scratch/RNA-Seq_example/ATH/trim
cd ~/scratch/RNA-Seq_example/ATH
nano trim.sh
#!/bin/bash
#SBATCH --job-name=trim_ATH
#SBATCH --cpus-per-task=2
#SBATCH --time=2-15:00:00
#SBATCH --mem=16g
#SBATCH --mail-type=ALL
#SBATCH --mail-user=<YOUR_EMAIL>
#SBATCH -o trim.out
#SBATCH --account=cpu-s5-bch709-5
#SBATCH --partition=cpu-core-0
trim_galore --paired --three_prime_clip_R1 5 --three_prime_clip_R2 5 --cores 2 --max_n 40 --gzip -o ~/scratch/RNA-Seq_example/ATH/trim --basename SRR1761506 raw_data/SRR1761506_1.fastq.gz raw_data/SRR1761506_2.fastq.gz --fastqc
trim_galore --paired --three_prime_clip_R1 5 --three_prime_clip_R2 5 --cores 2 --max_n 40 --gzip -o ~/scratch/RNA-Seq_example/ATH/trim --basename SRR1761507 raw_data/SRR1761507_1.fastq.gz raw_data/SRR1761507_2.fastq.gz --fastqc
trim_galore --paired --three_prime_clip_R1 5 --three_prime_clip_R2 5 --cores 2 --max_n 40 --gzip -o ~/scratch/RNA-Seq_example/ATH/trim --basename SRR1761508 raw_data/SRR1761508_1.fastq.gz raw_data/SRR1761508_2.fastq.gz --fastqc
trim_galore --paired --three_prime_clip_R1 5 --three_prime_clip_R2 5 --cores 2 --max_n 40 --gzip -o ~/scratch/RNA-Seq_example/ATH/trim --basename SRR1761509 raw_data/SRR1761509_1.fastq.gz raw_data/SRR1761509_2.fastq.gz --fastqc
trim_galore --paired --three_prime_clip_R1 5 --three_prime_clip_R2 5 --cores 2 --max_n 40 --gzip -o ~/scratch/RNA-Seq_example/ATH/trim --basename SRR1761510 raw_data/SRR1761510_1.fastq.gz raw_data/SRR1761510_2.fastq.gz --fastqc
trim_galore --paired --three_prime_clip_R1 5 --three_prime_clip_R2 5 --cores 2 --max_n 40 --gzip -o ~/scratch/RNA-Seq_example/ATH/trim --basename SRR1761511 raw_data/SRR1761511_1.fastq.gz raw_data/SRR1761511_2.fastq.gz --fastqc
Submit trimming
squeue -u $(whoami)
sbatch --dependency=afterok:######## trim.sh
Explain the command
This command runs **Trim Galore**, a tool used for trimming adapters and low-quality sequences from high-throughput sequencing data, with specific parameters for paired-end RNA-Seq reads. Here’s a detailed breakdown:
1. **`trim_galore --paired`**: This specifies that the input data consists of paired-end reads. Trim Galore will process both forward and reverse reads together, ensuring paired-end compatibility after trimming.
2. **`--three_prime_clip_R1 5` and `--three_prime_clip_R2 5`**: These options clip (remove) 5 bases from the 3' end of both reads in the pair. **`_R1`** applies to the first read and **`_R2`** applies to the second read, which can help remove low-quality or unwanted bases at the end of each read.
3. **`--cores 2`**: This specifies the number of cores (CPUs) to use, allowing Trim Galore to run with 2 parallel threads for faster processing.
4. **`--max_n 40`**: This sets the maximum number of ambiguous bases (`N`) allowed per read. Reads with more than 40 `N` bases will be discarded, helping improve the quality of the data.
5. **`--gzip`**: This option compresses the output files in **GZIP** format, saving space and making downstream handling more efficient.
6. **`-o ~/scratch/RNA-Seq_example/ATH/trim`**: This specifies the output directory for the processed files. Here, the results will be saved in `~/scratch/RNA-Seq_example/ATH/trim`.
7. **`--basename SRR1761511`**: This sets the base name for the output files, which will start with **SRR1761511**. This is useful for organizing results by sample name.
8. **`raw_data/SRR1761511_1.fastq.gz raw_data/SRR1761511_2.fastq.gz`**: These are the input files: the paired-end FASTQ files (forward and reverse) that will be trimmed.
9. **`--fastqc`**: This tells Trim Galore to run **FastQC** on the trimmed reads, generating a quality control report. FastQC provides information on read quality, adapter content, and other metrics.
### In summary:
This command will trim adapters and low-quality bases from paired-end RNA-Seq reads in **SRR1761511**, remove excess ambiguous bases, clip 5 bases from the 3' ends of both reads, and save the results (compressed) to the specified output directory with **FastQC** quality reports. This helps prepare high-quality reads for further analysis.
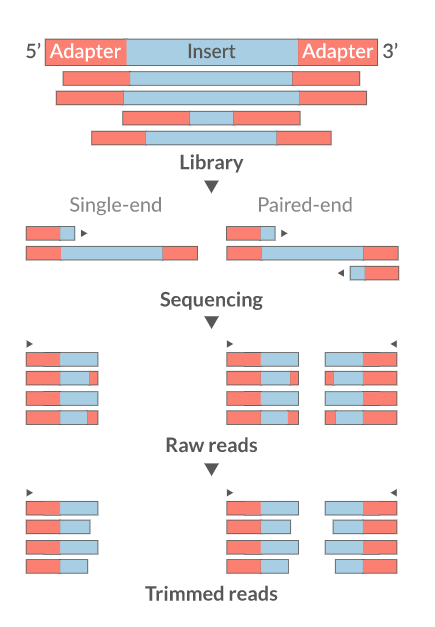
Trim the reads
- Trim IF necessary
- Synthetic bases can be an issue for SNP calling
- Insert size distribution may be more important for assemblers
- Trim/Clip/Filter reads
- Remove adapter sequences
- Trim reads by quality
- Sliding window trimming
- Filter by min/max read length
- Remove reads less than ~18nt
- Demultiplexing/Splitting
Cutadapt
fastp
Skewer
Prinseq
Trimmomatics
Trim Galore
Download reference file
cd ~/scratch/RNA-Seq_example/ATH
mkdir bam
mkdir reference
cd reference
wget https://www.arabidopsis.org/api/download-files/download?filePath=Genes/TAIR10_genome_release/TAIR10_chromosome_files/TAIR10_chr_all.fas.gz -O TAIR10_chr_all.fas.gz --no-check-certificate
wget https://www.arabidopsis.org/api/download-files/download?filePath=Genes/TAIR10_genome_release/TAIR10_gff3/TAIR10_GFF3_genes.gff -O TAIR10_GFF3_genes.gff --no-check-certificate
seqkit stats TAIR10_chr_all.fas.gz
Explain the command
This command uses **wget** to download a GFF3 file from the Arabidopsis Information Resource (TAIR) website. Here’s what each part does:
1. **`wget https://www.arabidopsis.org/api/download-files/download?filePath=Genes/TAIR10_genome_release/TAIR10_gff3/TAIR10_GFF3_genes.gff`**: This is the download command with the full URL of the TAIR10 GFF3 file. **GFF3** (General Feature Format, version 3) files are used for genome annotations and contain information about gene locations, exons, coding regions, and other genomic features.
2. **`-O TAIR10_GFF3_genes.gff`**: This option specifies the output filename. Without this option, `wget` would use the default name provided by the URL, which might be complex or include additional characters. **TAIR10_GFF3_genes.gff** is set as the filename to make it easier to reference.
3. **`--no-check-certificate`**: This option tells `wget` to ignore SSL certificate verification errors. It can be useful when a website’s SSL certificate is expired or not properly recognized. Here, it ensures the download proceeds without interruptions.
### Summary:
This command downloads the **TAIR10_GFF3_genes.gff** file from the TAIR database without SSL verification, saving it as **TAIR10_GFF3_genes.gff**. This GFF3 file will contain essential genome annotation data for Arabidopsis, commonly used in genomic analysis.
Convert GFF to GTF
cd ~/scratch/RNA-Seq_example/ATH/reference
gffread TAIR10_GFF3_genes.gff -T -F --keep-exon-attrs -o TAIR10_GFF3_genes.gtf
Explain the command
This command uses **gffread** to convert a **GFF3** file into a **GTF** file format. **GTF** (Gene Transfer Format) is similar to **GFF** but often preferred in certain bioinformatics tools, especially for transcript-based analyses. Here’s a breakdown of each component:
1. **`gffread TAIR10_GFF3_genes.gff`**: This specifies the input file in **GFF3** format, here **TAIR10_GFF3_genes.gff**.
2. **`-T`**: This option tells `gffread` to output the file in **GTF** format instead of **GFF3**.
3. **`-F`**: This forces the inclusion of features that might be incomplete or missing certain attributes (e.g., lacking start or stop codons). It ensures that all exons are included in the output.
4. **`--keep-exon-attrs`**: This option retains additional attributes associated with exon features, which are often discarded in standard conversions. Keeping exon attributes can be valuable for certain analyses where more detailed annotation is needed.
5. **`-o TAIR10_GFF3_genes.gtf`**: This specifies the output filename. Here, the GTF-formatted file will be saved as **TAIR10_GFF3_genes.gtf**.
### Summary:
This command converts the **TAIR10** genome annotation file from **GFF3** to **GTF** format, ensuring that all exons are retained along with their attributes, even if they lack some standard annotations. This output file, **TAIR10_GFF3_genes.gtf**, will be useful for downstream transcript-based analysis in pipelines or tools that prefer GTF format.
Create reference index
cd ~/scratch/RNA-Seq_example/ATH/reference
ls -algh
gunzip TAIR10_chr_all.fas.gz
nano index.sh
#!/bin/bash
#SBATCH --job-name=index_ATH
#SBATCH --cpus-per-task=12
#SBATCH --time=2-15:00:00
#SBATCH --mem=48g
#SBATCH --mail-type=all
#SBATCH --mail-user=<PLEASE CHANGE THIS TO YOUR EMAIL>
#SBATCH -o index.out # STDOUT & STDERR
#SBATCH --account=cpu-s5-bch709-5
#SBATCH --partition=cpu-core-0
STAR --runThreadN 48g --runMode genomeGenerate --genomeDir . --genomeFastaFiles ~/scratch/RNA-Seq_example/ATH/reference/TAIR10_chr_all.fas --sjdbGTFfile ~/scratch/RNA-Seq_example/ATH/reference/TAIR10_GFF3_genes.gtf --sjdbOverhang 99 --genomeSAindexNbases 12
Explain the command
This command uses **STAR** (Spliced Transcripts Alignment to a Reference) to generate a genome index, which is a critical step for efficiently aligning RNA-Seq reads to a reference genome. Here’s a detailed breakdown of each parameter:
1. **`STAR`**: This calls the STAR program, which is an RNA-Seq read aligner optimized for high accuracy and speed.
2. **`--runThreadN 48g`**: This option specifies the number of threads (CPUs) STAR should use. However, the argument should be an integer (like `48`) rather than `48g`. Assuming you intended to use 48 threads, the correct syntax would be `--runThreadN 48`.
3. **`--runMode genomeGenerate`**: This tells STAR to run in genome generation mode, which creates an index for the reference genome. This index is needed for subsequent alignment steps.
4. **`--genomeDir .`**: This sets the directory where the generated genome index files will be stored. Using `.` specifies the current directory.
5. **`--genomeFastaFiles ~/scratch/RNA-Seq_example/ATH/reference/TAIR10_chr_all.fas`**: This provides the path to the reference genome FASTA file. Here, **TAIR10_chr_all.fas** contains the reference sequences for **Arabidopsis thaliana**.
6. **`--sjdbGTFfile ~/scratch/RNA-Seq_example/ATH/reference/TAIR10_GFF3_genes.gtf`**: This specifies the path to the annotation file in **GTF** format (TAIR10_GFF3_genes.gtf). STAR uses this file to incorporate known splice junctions into the index, which improves alignment accuracy, especially for spliced reads in RNA-Seq data.
7. **`--sjdbOverhang 99`**: This defines the length of the sequence to be used for junctions. Ideally, it should be set to the length of the read minus 1. For instance, if the RNA-Seq reads are 100 bp, `sjdbOverhang` should be 99. This value helps STAR optimize the alignment of reads that span splice junctions.
8. **`--genomeSAindexNbases 12`**: This parameter controls the size of the suffix array index used by STAR. **12** is typical for a smaller genome (such as Arabidopsis) to balance between memory usage and indexing speed. Larger values reduce memory requirements but can slow down the alignment step slightly.
### Summary:
This command sets STAR to generate a genome index for **Arabidopsis thaliana** using 48 threads, based on the reference FASTA and GTF files. The generated index will be saved in the current directory and includes splice junction information, which is crucial for accurately mapping RNA-Seq reads that contain introns. This index will be used in future alignment steps to align RNA-Seq reads quickly and accurately to the genome.
Mapping the reads to genome index
cd ~/scratch/RNA-Seq_example/ATH/
ls -algh
nano align.sh
#!/bin/bash
#SBATCH --job-name=align_ATH
#SBATCH --cpus-per-task=8
#SBATCH --time=2-15:00:00
#SBATCH --mem=32g
#SBATCH --mail-type=all
#SBATCH --mail-user=<PLEASE CHANGE THIS TO YOUR EMAIL>
#SBATCH -o align.out # STDOUT & STDERR
#SBATCH --account=cpu-s5-bch709-5
#SBATCH --partition=cpu-core-0
STAR --runMode alignReads --runThreadN 8 --readFilesCommand zcat --outFilterMultimapNmax 10 --alignIntronMin 25 --alignIntronMax 10000 --genomeDir ~/scratch/RNA-Seq_example/ATH/reference/ --readFilesIn ~/scratch/RNA-Seq_example/ATH/trim/SRR1761506_val_1.fq.gz ~/scratch/RNA-Seq_example/ATH/trim/SRR1761506_val_2.fq.gz --outSAMtype BAM SortedByCoordinate --outFileNamePrefix ~/scratch/RNA-Seq_example/ATH/bam/SRR1761506.bam
STAR --runMode alignReads --runThreadN 8 --readFilesCommand zcat --outFilterMultimapNmax 10 --alignIntronMin 25 --alignIntronMax 10000 --genomeDir ~/scratch/RNA-Seq_example/ATH/reference/ --readFilesIn ~/scratch/RNA-Seq_example/ATH/trim/SRR1761507_val_1.fq.gz ~/scratch/RNA-Seq_example/ATH/trim/SRR1761507_val_2.fq.gz --outSAMtype BAM SortedByCoordinate --outFileNamePrefix ~/scratch/RNA-Seq_example/ATH/bam/SRR1761507.bam
STAR --runMode alignReads --runThreadN 8 --readFilesCommand zcat --outFilterMultimapNmax 10 --alignIntronMin 25 --alignIntronMax 10000 --genomeDir ~/scratch/RNA-Seq_example/ATH/reference/ --readFilesIn ~/scratch/RNA-Seq_example/ATH/trim/SRR1761508_val_1.fq.gz ~/scratch/RNA-Seq_example/ATH/trim/SRR1761508_val_2.fq.gz --outSAMtype BAM SortedByCoordinate --outFileNamePrefix ~/scratch/RNA-Seq_example/ATH/bam/SRR1761508.bam
STAR --runMode alignReads --runThreadN 8 --readFilesCommand zcat --outFilterMultimapNmax 10 --alignIntronMin 25 --alignIntronMax 10000 --genomeDir ~/scratch/RNA-Seq_example/ATH/reference/ --readFilesIn ~/scratch/RNA-Seq_example/ATH/trim/SRR1761509_val_1.fq.gz ~/scratch/RNA-Seq_example/ATH/trim/SRR1761509_val_2.fq.gz --outSAMtype BAM SortedByCoordinate --outFileNamePrefix ~/scratch/RNA-Seq_example/ATH/bam/SRR1761509.bam
STAR --runMode alignReads --runThreadN 8 --readFilesCommand zcat --outFilterMultimapNmax 10 --alignIntronMin 25 --alignIntronMax 10000 --genomeDir ~/scratch/RNA-Seq_example/ATH/reference/ --readFilesIn ~/scratch/RNA-Seq_example/ATH/trim/SRR1761510_val_1.fq.gz ~/scratch/RNA-Seq_example/ATH/trim/SRR1761510_val_2.fq.gz --outSAMtype BAM SortedByCoordinate --outFileNamePrefix ~/scratch/RNA-Seq_example/ATH/bam/SRR1761510.bam
STAR --runMode alignReads --runThreadN 8 --readFilesCommand zcat --outFilterMultimapNmax 10 --alignIntronMin 25 --alignIntronMax 10000 --genomeDir ~/scratch/RNA-Seq_example/ATH/reference/ --readFilesIn ~/scratch/RNA-Seq
Submit mapping
squeue -u $(whoami)
sbatch --dependency=afterok:########:####### align.sh
Explain the command
This command uses **STAR** in **alignment mode** to align paired-end RNA-Seq reads to a pre-built genome index. Here’s a detailed breakdown:
1. **`STAR --runMode alignReads`**: Specifies that STAR should run in **alignment mode**, which aligns RNA-Seq reads to the reference genome.
2. **`--runThreadN 8`**: Sets the number of threads to use (in this case, 8), which will speed up the alignment process by utilizing multiple CPU cores.
3. **`--readFilesCommand zcat`**: Instructs STAR to use `zcat` to decompress the input files since they are **GZIP** compressed (`.fq.gz` format).
4. **`--outFilterMultimapNmax 10`**: Sets the maximum number of loci a read can map to. If a read maps to more than 10 locations, it will be discarded. This helps control the level of ambiguity in alignment, especially in repetitive regions.
5. **`--alignIntronMin 25`**: Specifies the minimum allowed length for introns. STAR will ignore introns shorter than 25 bp, which reduces false alignments in smaller repetitive regions.
6. **`--alignIntronMax 10000`**: Sets the maximum allowed intron length to 10,000 bp, accommodating typical intron lengths found in **Arabidopsis**. This helps STAR avoid spurious alignments that would involve unusually long gaps.
7. **`--genomeDir ~/scratch/RNA-Seq_example/ATH/reference/`**: Specifies the directory containing the STAR genome index, created in the previous genome generation step.
8. **`--readFilesIn ~/scratch/RNA-Seq_example/ATH/trim/SRR1761506_val_1.fq.gz ~/scratch/RNA-Seq_example/ATH/trim/SRR1761506_val_2.fq.gz`**: These are the paths to the input FASTQ files for paired-end reads (forward and reverse), which have been trimmed and compressed.
9. **`--outSAMtype BAM SortedByCoordinate`**: Specifies that the output should be in **BAM** format and sorted by genomic coordinates. BAM is a binary, compressed format for alignment data, commonly used for downstream analysis.
10. **`--outFileNamePrefix ~/scratch/RNA-Seq_example/ATH/bam/SRR1761506.bam`**: Sets the output filename prefix, so the results will be saved with the prefix `SRR1761506.bam` in the specified BAM directory. STAR will automatically append additional information as needed.
### Summary:
This command aligns the trimmed, paired-end RNA-Seq reads for **SRR1761506** to the **Arabidopsis** reference genome, utilizing 8 threads. It decompresses the input files, filters out highly multimapping reads, and limits intron size for optimized mapping. The output is saved in sorted **BAM** format, ready for downstream analysis.
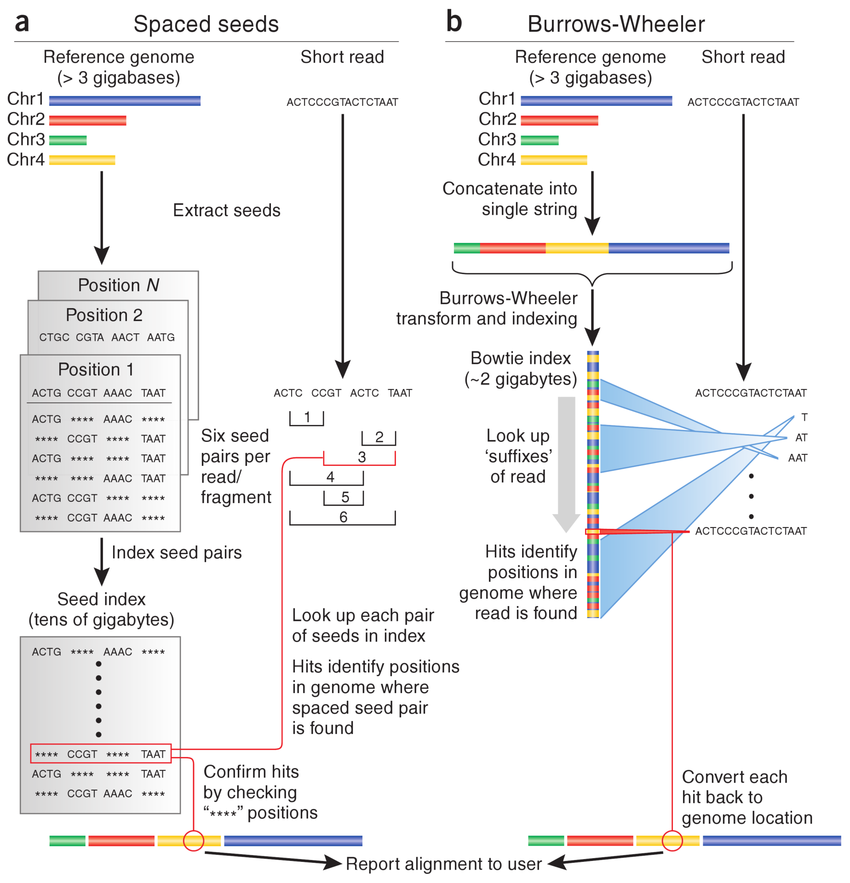
BW algorithm

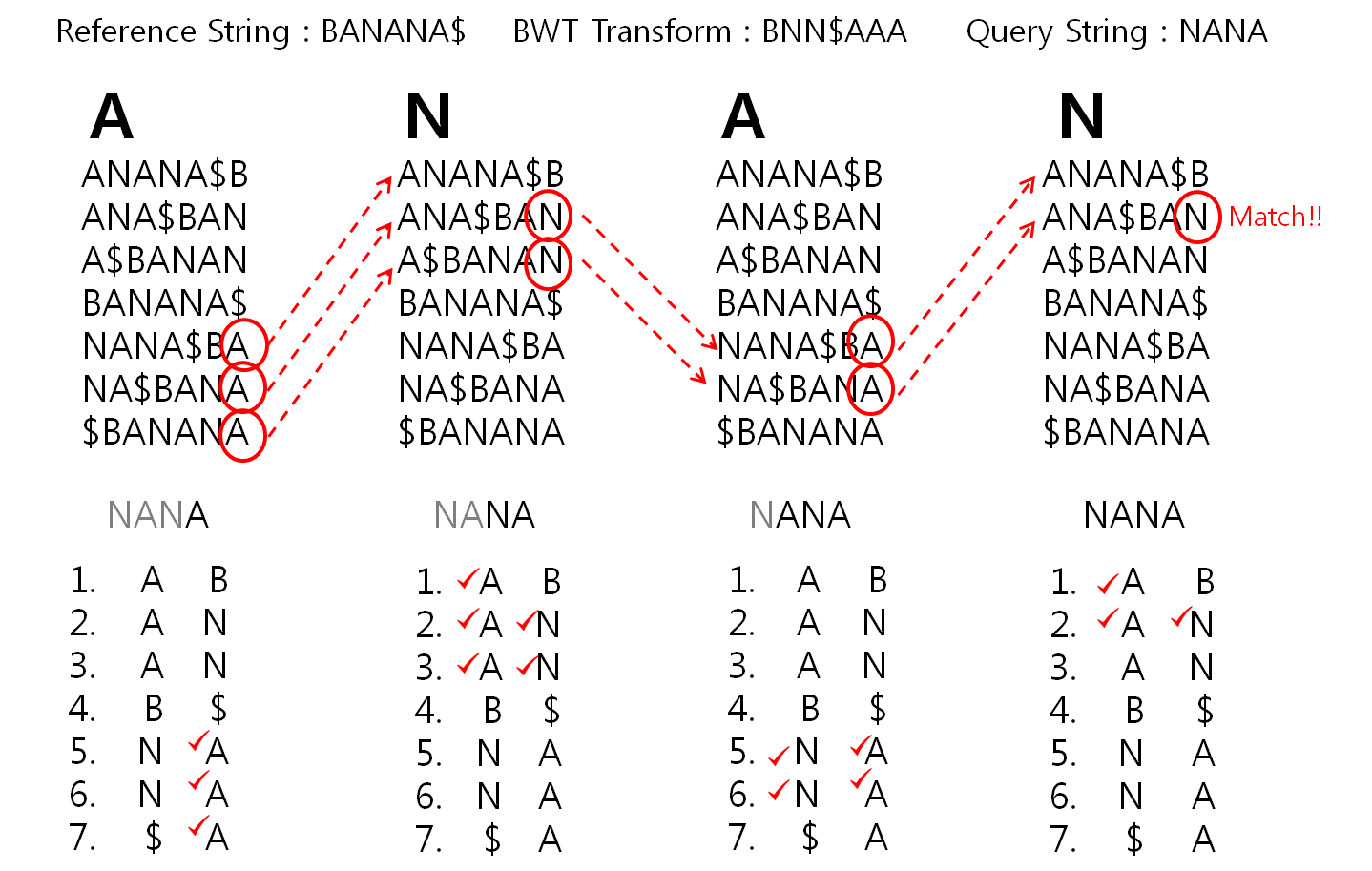
####################################
Mouse RNA-Seq
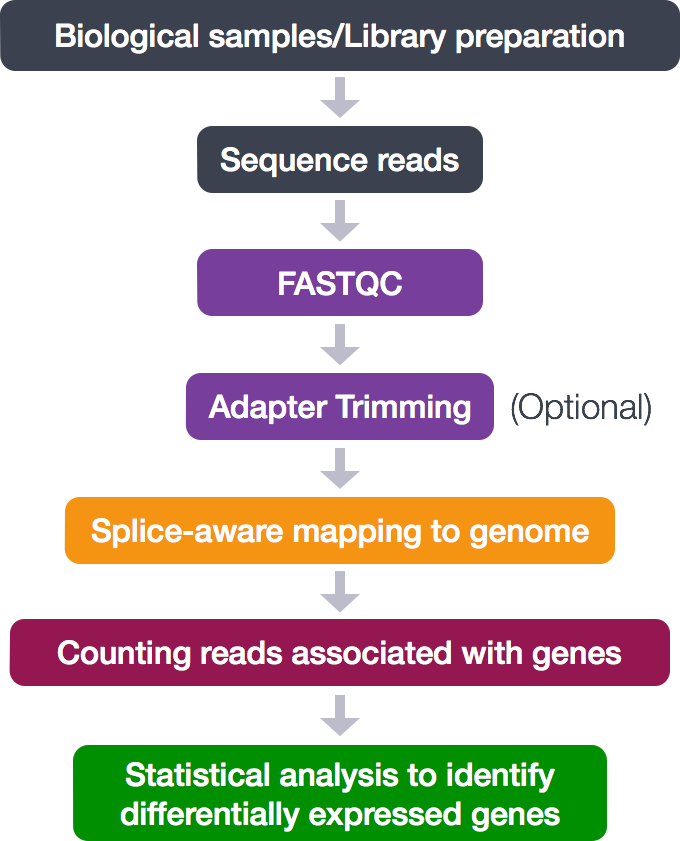
https://www.sciencedirect.com/science/article/pii/S2211124722011111
Benraiss A et al., “A TCF7L2-responsive suppression of both homeostatic and compensatory remyelination in Huntington disease mice.”, Cell Rep, 2022 Aug 30;40(9):111291
Working directory (Pronghorn)
echo $USER
cd /data/gpfs/assoc/bch709-5/students/${USER}
mkdir mouse
mkdir /data/gpfs/assoc/bch709-5/students/${USER}/mouse/fastq
mkdir /data/gpfs/assoc/bch709-5/students/${USER}/mouse/ref
mkdir /data/gpfs/assoc/bch709-5/students/${USER}/mouse/trim
mkdir /data/gpfs/assoc/bch709-5/students/${USER}/mouse/bam
mkdir /data/gpfs/assoc/bch709-5/students/${USER}/mouse/readcount
mkdir /data/gpfs/assoc/bch709-5/students/${USER}/mouse/DEG
Reference Download
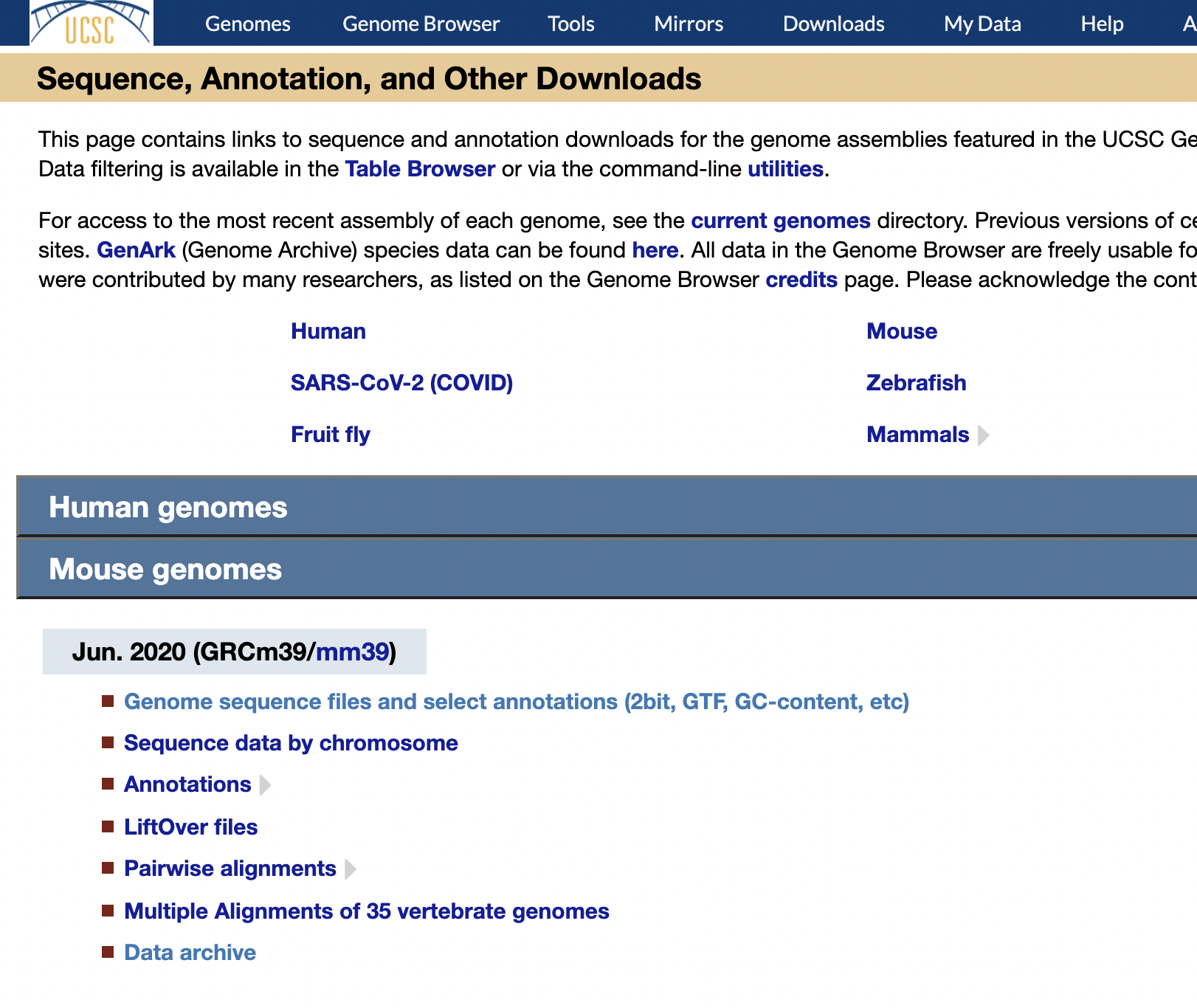
https://hgdownload.soe.ucsc.edu/downloads.html
### change working directory
cd /data/gpfs/assoc/bch709-5/students/${USER}/mouse/ref
### download
wget https://hgdownload.soe.ucsc.edu/goldenPath/mm39/bigZips/mm39.fa.gz
wget https://hgdownload.soe.ucsc.edu/goldenPath/mm39/bigZips/genes/refGene.gtf.gz
### decompress
gunzip mm39.fa.gz
gunzip refGene.gtf.gz
### index
nano index.sh
#!/bin/bash
#SBATCH --job-name=index_mouse
#SBATCH --cpus-per-task=12
#SBATCH --time=2-15:00:00
#SBATCH --mem=48g
#SBATCH --mail-type=all
#SBATCH --mail-user=<PLEASE CHANGE THIS TO YOUR EMAIL>
#SBATCH -o index.out # STDOUT & STDERR
#SBATCH --account=cpu-s5-bch709-5
#SBATCH --partition=cpu-core-0
STAR --runThreadN 48g --runMode genomeGenerate --genomeDir . --genomeFastaFiles /data/gpfs/assoc/bch709-5/students/${USER}/mouse/ref/mm39.fa --sjdbGTFfile /data/gpfs/assoc/bch709-5/students/${USER}/mouse/ref/refGene.gtf --sjdbOverhang 99 --genomeSAindexNbases 12
FASTQ file
cd /data/gpfs/assoc/bch709-5/students/${USER}/mouse/
### Link file (without copy)
ln -s /data/gpfs/assoc/bch709-5/students/Course_materials/mouse/fastq/* /data/gpfs/assoc/bch709-5/students/${USER}/mouse/fastq
ls /data/gpfs/assoc/bch709-5/students/${USER}/mouse/fastq
Create file list
cd /data/gpfs/assoc/bch709-5/students/${USER}/mouse/fastq
ls -1 *.gz | sed 's/_R.\.fastq\.gz//g' | sort -u > /data/gpfs/assoc/bch709-5/students/${USER}/mouse/filelist
cat /data/gpfs/assoc/bch709-5/students/${USER}/mouse/filelist
Regular expression
https://regex101.com/
Trim reads
trim_galore --paired --three_prime_clip_R1 [integer] --three_prime_clip_R2 [integer] --cores [integer] --max_n [integer] --fastqc --gzip -o /data/gpfs/assoc/bch709-5/students/${USER}/mouse/trim {READ_R1} {READ_R2}
Trim reads example
trim_galore --paired --three_prime_clip_R1 5 --three_prime_clip_R2 5 --cores 2 --max_n 40 --fastqc --gzip -o /data/gpfs/assoc/bch709-5/students/${USER}/mouse/trim {READ_R1} {READ_R2}
Prepare templet
cat /data/gpfs/assoc/bch709-5/students/Course_materials/mouse/run.sh | sed "s/\-\-cpus\-per\-task\=2/\-\-cpus\-per\-task\=4/g; s/\[NAME\]/Trim/g; s/\[youremail\]/${USER}\@unr.edu\,${USER}\@nevada.unr.edu/g" > /data/gpfs/assoc/bch709-5/students/${USER}/mouse/fastq/trim.sh
Edit templet
nano /data/gpfs/assoc/bch709-5/students/${USER}/mouse/fastq/trim.sh
Batch submission
# Check file list
cat ../filelist
# Loop file list
### Add Forward read to variable
### Add reverse read from forward read name substitution
for i in `cat ../filelist`
do
read1=${i}_R1.fastq.gz
read2=${read1//_R1.fastq.gz/_R2.fastq.gz}
echo $read1 $read2
done
# Loop file list
### add file name from variable to trim-galore
### merge trim-galore command and trim.sh
for i in `cat ../filelist`
do
read1=${i}_R1.fastq.gz
read2=${read1//_R1.fastq.gz/_R2.fastq.gz}
echo $read1 $read2
echo "trim_galore --paired --three_prime_clip_R1 5 --three_prime_clip_R2 5 --cores 2 --max_n 40 --fastqc --gzip -o /data/gpfs/assoc/bch709-5/students/${USER}/mouse/trim $read1 $read2" | cat trim.sh - > ${i}_trim.sh
done
### Batch submission
ls *.sh
ls -1 *.sh
### Loop *.sh printing
for i in `ls -1 *.sh`
do
echo $i
done
### Loop *.sh submission
for i in `ls -1 *.sh`
do
sbatch $i
done
Check submission
squeue -u ${USER}
Environment activation
conda activate BCH709_RNASeq
Copy files
cp /data/gpfs/assoc/bch709-5/students/Course_materials/mouse/ref/* /data/gpfs/assoc/bch709-5/students/${USER}/mouse/ref/
cp /data/gpfs/assoc/bch709-5/students/Course_materials/mouse/trim/* /data/gpfs/assoc/bch709-5/students/${USER}/mouse/trim/
RNA-Seq Alignment
#### Move to trim folder
cd /data/gpfs/assoc/bch709-5/students/${USER}/mouse/trim
#### Copy templet
cat /data/gpfs/assoc/bch709-5/students/Course_materials/mouse/run.sh | sed "s/16g/64g/g; s/\-\-cpus\-per\-task\=2/\-\-cpus\-per\-task\=4/g; s/\[NAME\]/Trim/g; s/\[youremail\]/${USER}\@unr.edu\,${USER}\@nevada.unr.edu/g" > /data/gpfs/assoc/bch709-5/students/${USER}/mouse/trim/mapping.sh
#### Edit templet
nano mapping.sh
Check output
ls -algh /data/gpfs/assoc/bch709-5/students/${USER}/mouse/trim
Output example
[FILENAME]_R1_val_1.fq.gz [FILENAME]_R2_val_2.fq.gz
STAR RNA-Seq alignment batch file test
for i in `cat ../filelist`
do
read1=${i}_R1_val_1.fq.gz
read2=${read1//_R1_val_1.fq.gz/_R2_val_2.fq.gz}
echo $read1 $read2
echo "STAR --runMode alignReads --runThreadN 4 --outFilterMultimapNmax 100 --alignIntronMin 25 --alignIntronMax 50000 --genomeDir /data/gpfs/assoc/bch709-5/students/${USER}/mouse/ref --readFilesCommand gunzip -c --readFilesIn /data/gpfs/assoc/bch709-5/students/${USER}/mouse/trim/${read1} /data/gpfs/assoc/bch709-5/students/${USER}/mouse/trim/${read2} --outSAMtype BAM SortedByCoordinate --outFileNamePrefix /data/gpfs/assoc/bch709-5/students/${USER}/mouse/bam/${i}.bam" | cat mapping.sh - > ${i}_mapping.sh
done
Job submission dependency on Mapping
for i in `ls -1 *_mapping.sh`
do
sbatch $i
done
Reads count
In the case of RNA-Seq, the features are typically genes, where each gene is considered here as the union of all its exons. Counting RNA-seq reads is complex because of the need to accommodate exon splicing. The common approach is to summarize counts at the gene level, by counting all reads that overlap any exon for each gene. In this method, gene annotation file from RefSeq or Ensembl is often used for this purpose. So far there are two major feature counting tools: featureCounts (Liao et al.) and htseq-count (Anders et al.)

Copy templet
mkdir /data/gpfs/assoc/bch709-5/students/${USER}/RNA-Seq_example/ATH/readcount cd /data/gpfs/assoc/bch709-5/students/${USER}/RNA-Seq_example/ATH
| cat /data/gpfs/assoc/bch709-5/students/Course_materials/mouse/run.sh | sed “s/16g/64g/g; s/--cpus-per-task=2/--cpus-per-task=4/g; s/[NAME]/Count/g; s/[youremail]/${USER}\@unr.edu\,${USER}\@nevada.unr.edu/g” > /data/gpfs/assoc/bch709-5/students/${USER}/RNA-Seq_example/ATH/count.sh |
FeatureCounts read bam file
mkdir /data/gpfs/assoc/bch709-5/students/${USER}/RNA-Seq_example/ATH/readcount/
cd /data/gpfs/assoc/bch709-5/students/${USER}/RNA-Seq_example/ATH/raw_data
ls -1 *.gz | sed 's/_.*//g' | sort -u > /data/gpfs/assoc/bch709-5/students/${USER}/RNA-Seq_example/ATH/filelist
cd /data/gpfs/assoc/bch709-5/students/${USER}/RNA-Seq_example/ATH/bam
ls -1 *.sortedByCoord.out.bam
ls -1 *.sortedByCoord.out.bam| tr '\n' ' '
cp /data/gpfs/assoc/bch709-5/students/Course_materials/ATH/reference/* /data/gpfs/assoc/bch709-5/students/${USER}/RNA-Seq_example/ATH/reference/TAIR10_GFF3_genes.gtf
###########
featureCounts -o [output] -T [threads] -Q 1 -p -M -g gene_id -a [GTF] [BAMs]
Edit file
#paste this to count.sh
featureCounts -o /data/gpfs/assoc/bch709-5/students/${USER}/RNA-Seq_example/ATH/readcount/ATH_featucount -T 4 -Q 1 -p -M -g gene_id -a /data/gpfs/assoc/bch709-5/students/${USER}/RNA-Seq_example/ATH/reference/TAIR10_GFF3_genes.gtf $(for i in `cat /data/gpfs/assoc/bch709-5/students/${USER}/RNA-Seq_example/ATH/filelist`; do echo ${i}.bamAligned.sortedByCoord.out.bam| tr '\n' ' ';done)
nano count.sh
RNA-Seq report
How to make a report?

MultiQC
cd /data/gpfs/assoc/bch709-5/students/${USER}/RNA-Seq_example/ATH
multiqc --pdf -n test .
Slurm provides resource management for the processors allocated to a job, so that multiple job steps can be simultaneously submitted and queued until there are available resources within the job’s allocation. ############
FeatureCounts execute location
#### Move to trim folder
cd /data/gpfs/assoc/bch709-5/students/${USER}/mouse/bam
ls -1 *.bam
#### Copy templet
cp /data/gpfs/assoc/bch709-5/students/Course_materials/mouse/run.sh /data/gpfs/assoc/bch709-5/students/${USER}/mouse/bam/count.sh
sed -i "s/16g/64g/g; s/\-\-cpus\-per\-task\=2/\-\-cpus\-per\-task\=4/g; s/\[NAME\]/Count/g; s/\[youremail\]/${USER}\@unr.edu\,${USER}\@nevada.unr.edu/g" /data/gpfs/assoc/bch709-5/students/${USER}/mouse/bam/count.sh
FeatureCounts read bam file
cd /data/gpfs/assoc/bch709-5/students/${USER}/mouse/bam
ls -1 *.sortedByCoord.out.bam
ls -1 *.sortedByCoord.out.bam| tr '\n' ' '
Edit templet
nano count.sh
#paste this to count.sh
featureCounts -o /data/gpfs/assoc/bch709-5/students/${USER}//mouse/readcount/featucount -T 4 -Q 1 -p -M -g gene_id -a /data/gpfs/assoc/bch709-5/students/${USER}/mouse/ref/refGene.gtf $(for i in `cat /data/gpfs/assoc/bch709-5/students/${USER}/mouse/filelist`; do echo ${i}.bamAligned.sortedByCoord.out.bam| tr '\n' ' ';done)
Job submission dependency
squeue --noheader --format %i --user ${USER}
Submit
# Get the list of job IDs as a colon-separated string
jobid=$(squeue --noheader --format %i --user ${USER} | tr '\n' ':' | sed 's/:$//')
# Check if the jobid variable is empty
if [[ -n "$jobid" ]]; then
# If not empty, schedule the job with dependencies
sbatch --dependency=afterany:${jobid} count.sh
else
# If empty, schedule the job without dependencies
sbatch count.sh
fi
local Mac to Downloads
echo "setopt nonomatch" >> ~/.zshrc
Conda environment
mamba create -n RNASeq_postanalysis -y -c bioconda -c conda-forge -c r r trinity multiqc=1.9 samtools r-fastcluster=1.1.25 bioconductor-ctc bioconductor-deseq2 bioconductor-biobase=2.40.0 bioconductor-qvalue bioconductor-limma bioconductor-edger bioconductor-genomeinfodb bioconductor-deseq2 bioconductor-genomeinfodbdata r-rcurl
conda activate RNASeq_postanalysis
WORKTING PATH
cd /data/gpfs/assoc/bch709-5/students/${USER}/RNA-Seq_example/ATH/readcount
Reads count
In the case of RNA-Seq, the features are typically genes, where each gene is considered here as the union of all its exons. Counting RNA-seq reads is complex because of the need to accommodate exon splicing. The common approach is to summarize counts at the gene level, by counting all reads that overlap any exon for each gene. In this method, gene annotation file from RefSeq or Ensembl is often used for this purpose. So far there are two major feature counting tools: featureCounts (Liao et al.) and htseq-count (Anders et al.)
head ATH_featucount
cut -f1,7- ATH_featucount | egrep -v "#" | sed 's/\Aligned\.sortedByCoord\.out\.bam//g; s/\.bam//g' >> ATH.featureCount_count_only.cnt
Go to DEG
head ATH_featucount
head ATH.featureCount_count_only.cnt
mkdir /data/gpfs/assoc/bch709-5/students/${USER}/RNA-Seq_example/ATH/DEG
cp ATH.featureCount* ../DEG
cd /data/gpfs/assoc/bch709-5/students/${USER}/RNA-Seq_example/ATH/DEG
ls
Data list
| Sample information | Run |
|---|---|
| WT_rep1 | SRR1761506 |
| WT_rep2 | SRR1761507 |
| WT_rep3 | SRR1761508 |
| ABA_rep1 | SRR1761509 |
| ABA_rep2 | SRR1761510 |
| ABA_rep3 | SRR1761511 |
sample files
nano samples.txt
WT<TAB>SRR1761506
WT<TAB>SRR1761507
WT<TAB>SRR1761508
ABA<TAB>SRR1761509
ABA<TAB>SRR1761510
ABA<TAB>SRR1761511
sed -i 's/<TAB>/\t/g' samples.txt
PtR (Quality Check Your Samples and Biological Replicates)
Once you’ve performed transcript quantification for each of your biological replicates, it’s good to examine the data to ensure that your biological replicates are well correlated, and also to investigate relationships among your samples. If there are any obvious discrepancies among your sample and replicate relationships such as due to accidental mis-labeling of sample replicates, or strong outliers or batch effects, you’ll want to identify them before proceeding to subsequent data analyses (such as differential expression).
PtR --matrix ATH.featureCount_count_only.cnt --samples samples.txt --CPM --log2 --min_rowSums 10 --sample_cor_matrix --compare_replicates
WT.rep_compare.pdf
ABA.rep_compare.pdf
DEG calculation
conda activate RNASeq_postanalysis
mamba install -c conda-forge -c anaconda pandas
cd /data/gpfs/assoc/bch709-5/students/${USER}/RNA-Seq_example/ATH/DEG
run_DE_analysis.pl --matrix ATH.featureCount_count_only.cnt --method DESeq2 --samples_file samples.txt --output rnaseq
DEG output
cd rnaseq
ATH.featureCount_count_only.cnt.ABA_vs_WT.DESeq2.count_matrix
ATH.featureCount_count_only.cnt.ABA_vs_WT.DESeq2.DE_results
ATH.featureCount_count_only.cnt.ABA_vs_WT.DESeq2.DE_results.MA_n_Volcano.pdf
ATH.featureCount_count_only.cnt.ABA_vs_WT.DESeq2.Rscript
TPM and FPKM calculation
cd /data/gpfs/assoc/bch709-5/students/${USER}/RNA-Seq_example/ATH/readcount
cut -f1,6- ATH_featucount | egrep -v "#" | sed 's/\Aligned\.sortedByCoord\.out\.bam//g; s/\.bam//g' > ATH.featureCount_count_length.cnt
cp ATH.featureCount* ../DEG
cd /data/gpfs/assoc/bch709-5/students/${USER}/RNA-Seq_example/ATH/DEG
cp /data/gpfs/assoc/bch709-5/students/Course_materials/script/tpm_raw_exp_calculator.py .
python tpm_raw_exp_calculator.py -count ATH.featureCount_count_length.cnt
TPM and FPKM calculation output
ATH.featureCount_count_length.cnt.fpkm.xls
ATH.featureCount_count_length.cnt.fpkm.tab
ATH.featureCount_count_length.cnt.tpm.xls
ATH.featureCount_count_length.cnt.tpm.tab
DEG subset
cd /data/gpfs/assoc/bch709-5/students/wyim/RNA-Seq_example/ATH/DEG/rnaseq
analyze_diff_expr.pl --samples /data/gpfs/assoc/bch709-5/students/${USER}/RNA-Seq_example/ATH/DEG/samples.txt --matrix /data/gpfs/assoc/bch709-5/students/${USER}/RNA-Seq_example/ATH/DEG/ATH.featureCount_count_length.cnt.tpm.tab -P 0.01 -C 2 --output ATH
analyze_diff_expr.pl --samples /data/gpfs/assoc/bch709-5/students/${USER}/RNA-Seq_example/ATH/DEG/samples.txt --matrix /data/gpfs/assoc/bch709-5/students/${USER}/RNA-Seq_example/ATH/DEG/ATH.featureCount_count_length.cnt.tpm.tab -P 0.05 -C 1 --output ATH
DEG output
ATH.matrix.log2.centered.sample_cor_matrix.pdf
ATH.matrix.log2.centered.genes_vs_samples_heatmap.pdf
ATH.featureCount_count_only.cnt.ABA_vs_WT.DESeq2.DE_results.P0.01_C2.ABA-UP.subset
ATH.featureCount_count_only.cnt.ABA_vs_WT.DESeq2.DE_results.P0.01_C2.WT-UP.subset
ATH.featureCount_count_only.cnt.ABA_vs_WT.DESeq2.DE_results.P0.01_C2.DE.subset
ATH.featureCount_count_only.cnt.ABA_vs_WT.DESeq2.DE_results.P0.01_C1.ABA-UP.subset
ATH.featureCount_count_only.cnt.ABA_vs_WT.DESeq2.DE_results.P0.01_C1.WT-UP.subset
ATH.featureCount_count_only.cnt.ABA_vs_WT.DESeq2.DE_results.P0.01_C1.DE.subset
Mouse DEG
Activate environment
conda activate RNASeq_postanalysis
WORKTING PATH
cd /data/gpfs/assoc/bch709-5/students/${USER}/mouse/readcount
Clean sample name
### ls /data/gpfs/assoc/bch709-5/students/${USER}/mouse/readcount
cut -f1,7- mouse_featurecount | egrep -v "#" | sed 's/\Aligned\.sortedByCoord\.out\.bam//g; s/\.bam//g' > mouse_featurecount_only.cnt
cut -f1,6- mouse_featurecount | egrep -v "#" | sed 's/\Aligned\.sortedByCoord\.out\.bam//g; s/\.bam//g' > mouse_featurecount_length.cnt
Copy read count to DEG folder
cp /data/gpfs/assoc/bch709-5/students/${USER}/mouse/readcount/mouse_* /data/gpfs/assoc/bch709-5/students/${USER}/mouse/DEG/
Go to DEG folder
cd /data/gpfs/assoc/bch709-5/students/${USER}/mouse/DEG/
sample files
nano samples.txt
WT<TAB>R62_WT_Rep2
WT<TAB>R62_WT_Rep3
WT<TAB>R62_WT_Rep4
WT<TAB>R62_WT_Rep5
Glial<TAB>R62_Glial_Rep1
Glial<TAB>R62_Glial_Rep2
Glial<TAB>R62_Glial_Rep3
Glial<TAB>R62_Glial_Rep4
Glial<TAB>R62_Glial_Rep5
sed -i 's/<TAB>/\t/g' samples.txt
PtR (Quality Check Your Samples and Biological Replicates)
cd /data/gpfs/assoc/bch709-5/students/${USER}/mouse/DEG/
PtR --matrix mouse_featurecount_only.cnt --samples samples.txt --CPM --log2 --min_rowSums 10 --sample_cor_matrix --compare_replicates
PtR download on local
scp [YOURID]@pronghorn.rc.unr.edu:/data/gpfs/assoc/bch709-5/students/${USER}/mouse/DEG/*.pdf .
DEG calculation
cd /data/gpfs/assoc/bch709-5/students/${USER}/mouse/DEG
run_DE_analysis.pl --matrix mouse_featurecount_only.cnt --method DESeq2 --samples_file samples.txt --output rnaseq
DEG output
cd rnaseq
mouse_featurecount_only.cnt.Glial_vs_WT.DESeq2.count_matrix
mouse_featurecount_only.cnt.Glial_vs_WT.DESeq2.DE_results
mouse_featurecount_only.cnt.Glial_vs_WT.DESeq2.DE_results.MA_n_Volcano.pdf
mouse_featurecount_only.cnt.Glial_vs_WT.DESeq2.Rscript
TPM and FPKM calculation
TPM/FPKM calculation
cp /data/gpfs/assoc/bch709-5/students/Course_materials/script/tpm_raw_exp_calculator.py .
python tpm_raw_exp_calculator.py -count mouse_featurecount_length.cnt
TPM and FPKM calculation output
mouse_featurecount_length.cnt.fpkm.xls
mouse_featurecount_length.cnt.fpkm.tab
mouse_featurecount_length.cnt.tpm.xls
mouse_featurecount_length.cnt.tpm.tab
DEG subset
cd /data/gpfs/assoc/bch709-5/students/wyim/mouse/DEG/rnaseq
analyze_diff_expr.pl --samples /data/gpfs/assoc/bch709-5/students/wyim/mouse/DEG/samples.txt --matrix /data/gpfs/assoc/bch709-5/students/wyim/mouse/DEG/mouse_featurecount_length.cnt.tpm.tab -P 0.01 -C 2 --output mouse
analyze_diff_expr.pl --samples /data/gpfs/assoc/bch709-5/students/wyim/mouse/DEG/samples.txt --matrix /data/gpfs/assoc/bch709-5/students/wyim/mouse/DEG/mouse_featurecount_length.cnt.tpm.tab -P 0.01 -C 2 --output mouse
DEG output
mouse_featurecount_only.cnt.Glial_vs_WT.DESeq2.count_matrix
mouse_featurecount_only.cnt.Glial_vs_WT.DESeq2.DE_results
mouse_featurecount_only.cnt.Glial_vs_WT.DESeq2.DE_results.MA_n_Volcano.pdf
mouse_featurecount_only.cnt.Glial_vs_WT.DESeq2.DE_results.P0.01_C2.DE.subset
mouse_featurecount_only.cnt.Glial_vs_WT.DESeq2.DE_results.P0.01_C2.Glial-UP.subset
mouse_featurecount_only.cnt.Glial_vs_WT.DESeq2.DE_results.P0.01_C2.WT-UP.subset
mouse_featurecount_only.cnt.Glial_vs_WT.DESeq2.DE_results.samples
mouse_featurecount_only.cnt.Glial_vs_WT.DESeq2.Rscript
Expression normalization based on transcript length
FPKM
Fragments per Kilobase of transcript per million mapped reads
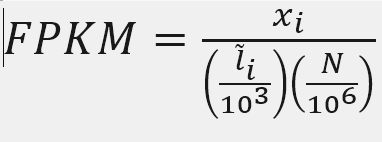
X = mapped reads count N = number of reads L = Length of transcripts
cd /data/gpfs/assoc/bch709-5/students/wyim/mouse/DEG/
head mouse_featurecount_length.cnt
awk 'NR > 1 {sum += $3} END {print sum}' mouse_featurecount_length.cnt
egrep Xkr4 /data/gpfs/assoc/bch709-5/students/wyim/mouse/DEG/rnaseq/rnaseqmouse_featurecount_length.cnt
X = 718
Number_Reads_mapped = 87203201
Length = 3634
fpkm= X*(1000/Length)*(1000000/Number_Reads_mapped)
fpkm
quit()
ten to the ninth power = 10**9
fpkm=X/(Number_Reads_mapped*Length)*10**9
fpkm
quit()
check th FPKM file
cd /data/gpfs/assoc/bch709-5/students/wyim/mouse/DEG/
egrep Xkr4 mouse_featurecount_length.cnt.fpkm.tab
TPM
Transcripts Per Million
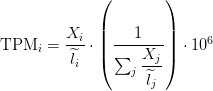
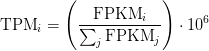
TPM calculation from reads count
cd /data/gpfs/assoc/bch709-5/students/wyim/mouse/DEG
awk 'NR > 1 {sum += $3/$2} END {print sum}' mouse_featurecount_length.cnt
sum_count_per_length = 32811.8
Length = 3634
X = 718
TPM = (X/Length)*(1/sum_count_per_length )*10**6
TPM calculation from FPKM
awk ‘NR > 1 {sum += $2} END {print sum}’ mouse_featurecount_length.cnt.fpkm.tab
FPKM = 2.265724465514574
SUM_FPKM = 376268
TPM=(FPKM/SUM_FPKM)*10**6
TPM
quit()
Paper read
Li et al., 2010, RSEM Dillies et al., 2013
DEG subset
cd /data/gpfs/assoc/bch709-5/students/wyim/mouse/DEG/rnaseq
analyze_diff_expr.pl --samples /data/gpfs/assoc/bch709-5/students/${USER}/mouse/DEG/samples.txt --matrix /data/gpfs/assoc/bch709-5/students/${USER}/mouse/DEG/mouse_featurecount_length.cnt.tpm.tab -P 0.01 -C 2 --output mouse_RNASEQ_P001_C2
analyze_diff_expr.pl --samples /data/gpfs/assoc/bch709-5/students/${USER}/mouse/DEG/samples.txt --matrix /data/gpfs/assoc/bch709-5/students/${USER}/mouse/DEG/mouse_featurecount_length.cnt.tpm.tab -P 0.05 -C 1 --output mouse_RNASEQ_P001_C2
DEG Subset to list
for i in *.subset; do
NAME=$(basename "${i}" .subset)
egrep -v sample "${i}" | cut -f 1 > "${NAME}.txt"
done
#This will generate .txt files for each .subset file in the directory, excluding lines with "sample" and retaining only the first column.
DEG download
scp ..
Functional analysis • GO
Gene enrichment analysis (Hypergeometric test) Gene set enrichment analysis (GSEA) Gene ontology / Reactome databases
Gene Ontology
Gene Ontology project is a major bioinformatics initiative Gene ontology is an annotation system The project provides the controlled and consistent vocabulary of terms and gene product annotations, i.e. terms occur only once, and there is a dictionary of allowed words GO describes how gene products behave in a cellular context A consistent description of gene products attributes in terms of their associated biological processes, cellular components and molecular functions in a species-independent manner Each GO term consists of a unique alphanumerical identifier, a common name, synonyms (if applicable), and a definition Each term is assigned to one of the three ontologies Terms have a textual definition When a term has multiple meanings depending on species, the GO uses a “sensu” tag to differentiate among them (trichome differentiation (sensu Magnoliophyta)
hypergeometric test
The hypergeometric distribution is the lesser-known cousin of the binomial distribution, which describes the probability of k successes in n draws with replacement. The hypergeometric distribution describes probabilities of drawing marbles from the jar without putting them back in the jar after each draw. The hypergeometric probability mass function is given by (using the original variable convention)
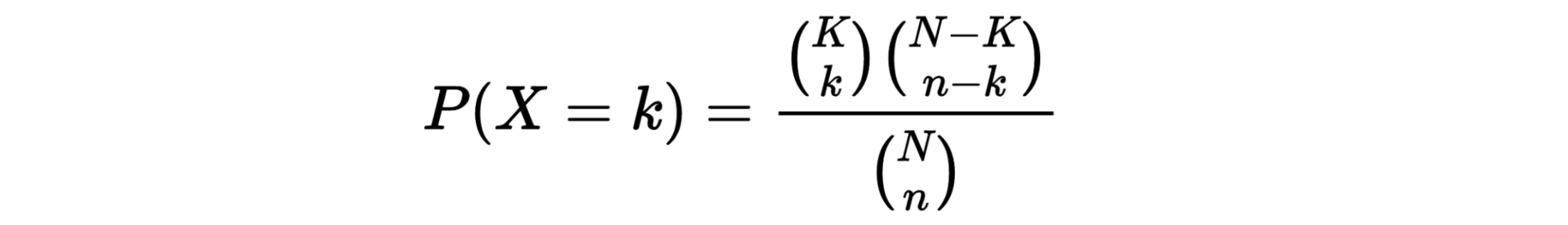

FWER
The FWER for the other tests is computed in the same way: the gene-associated variables (scores or counts) are permuted while the annotations of genes to GO-categories stay fixed. Then the statistical tests are evaluated again for every GO-category.
Hypergeometric Test Example 1
Suppose we randomly select 2 cards without replacement from an ordinary deck of playing cards. What is the probability of getting exactly 2 cards you want (i.e., Ace or 10)?
Solution: This is a hypergeometric experiment in which we know the following:
N = 52; since there are 52 cards in a deck. k = 16; since there are 16 Ace or 10 cards in a deck. n = 2; since we randomly select cards from the deck. x = 2; since 2 of the cards we select are red. We plug these values into the hypergeometric formula as follows:
h(x; N, n, k) = [ kCx ] [ N-kCn-x ] / [ NCn ]
h(2; 52, 2, 16) = [ 16C2 ] [ 48C1 ] / [ 52C2 ]
h(2; 52, 2, 16) = [ 325 ] [ 1 ] / [ 1,326 ]
h(2; 52, 2, 16) = 0.0904977
Thus, the probability of randomly selecting 2 Ace or 10 cards is 9%
| category | probability |
|---|---|
| probability mass f | 0.09049773755656108597285 |
| lower cumulative P | 1 |
| upper cumulative Q | 0.09049773755656108597285 |
| Expectation | 0.6153846153846153846154 |
Hypergeometric Test Example 2
Suppose we have 30 DEGs in human genome (200). What is the probability of getting 10 oncogene?
An oncogene is a gene that has the potential to cause cancer.
Solution: This is a hypergeometric experiment in which we know the following:
N = 200; since there are 200 genes in human genome k = 10; since there are 10 oncogenes in human n = 30; since 30 DEGs x = 5; since 5 of the oncogenes in DEGs.
We plug these values into the hypergeometric formula as follows:
h(x; N, n, k) = [ kCx ] [ N-kCn-x ] / [ NCn ]
h(5; 200, 30, 10) = [ 10C5 ] [ 190C25 ] / [ 200C30 ]
h(5; 200, 30, 10) = [ 252 ] [ 11506192278177947613740456466942 ] / [ 409681705022127773530866523638950880 ]
h(5; 200, 30, 10) = 0.007078
Thus, the probability of oncogene 0.7%.
hypergeometry.png
hypergeometric distribution value
| category | probability |
|---|---|
| probability mass f | 0.0070775932109153651831923063371216961166297 |
| lower cumulative P | 0.99903494867072865323201131115533112651846 |
| upper cumulative Q | 0.0080426445401867119511809951817905695981658 |
| Expectation | 1.5 |
False Discovery Rate (FDR) q-value
The false discovery rate (FDR) is a method of conceptualizing the rate of type I errors in null hypothesis testing when conducting multiple comparisons. FDR-controlling procedures are designed to control the expected proportion of “discoveries” (rejected null hypotheses) that are false (incorrect rejections).
- Benjamini–Yekutieli
- Benjamini–Hochberg
- Bonferroni-Selected–Bonferroni
- Bonferroni and Sidak
MetaScape
http://metascape.org/gp/index.html
REViGO
http://revigo.irb.hr/revigo.jsp
cleverGO
http://www.tartaglialab.com/GO_analyser/tutorial
DAVID
https://david.ncifcrf.gov/
Araport
http://araport.org
Paper read
Fu, Yu, et al. “Elimination of PCR duplicates in RNA-seq and small RNA-seq using unique molecular identifiers.” BMC genomics 19.1 (2018): 531 Parekh, Swati, et al. “The impact of amplification on differential expression analyses by RNA-seq.” Scientific reports 6 (2016): 25533 Klepikova, Anna V., et al. “Effect of method of deduplication on estimation of differential gene expression using RNA-seq.” PeerJ 5 (2017): e3091
Human RNA-Seq
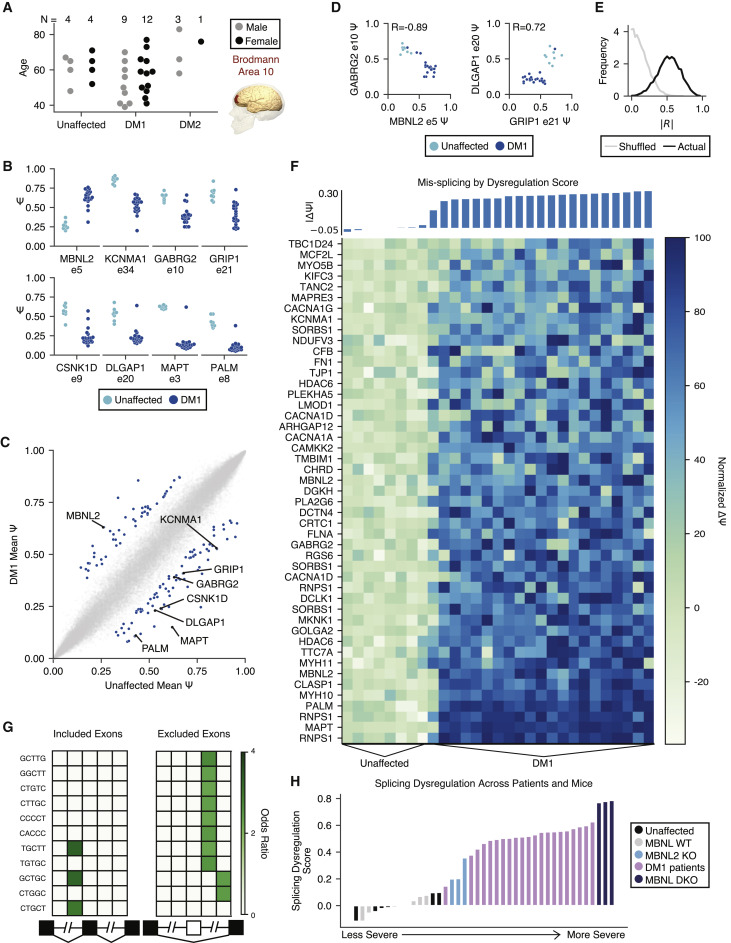
Transcriptome alterations in myotonic dystrophy frontal cortex
Environment activation
conda activate BCH709_RNASeq
Working directory (Pronghorn)
echo $USER
cd /data/gpfs/assoc/bch709-5/students/${USER}
mkdir human
mkdir /data/gpfs/assoc/bch709-5/students/${USER}/human/fastq
mkdir /data/gpfs/assoc/bch709-5/students/${USER}/human/ref
mkdir /data/gpfs/assoc/bch709-5/students/${USER}/human/trim
mkdir /data/gpfs/assoc/bch709-5/students/${USER}/human/bam
mkdir /data/gpfs/assoc/bch709-5/students/${USER}/human/readcount
mkdir /data/gpfs/assoc/bch709-5/students/${USER}/human/DEG
Reference Download
https://www.ncbi.nlm.nih.gov/genome/guide/human/
### change working directory
cd /data/gpfs/assoc/bch709-5/students/${USER}/human/ref
### download
wget https://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/genes/hg38.refGene.gtf.gz
wget https://ftp.ncbi.nlm.nih.gov/refseq/H_sapiens/annotation/GRCh38_latest/refseq_identifiers/GRCh38_latest_genomic.fna.gz
### decompress
gunzip GRCh38_latest_genomic.fna.gz
gunzip hg38.refGene.gtf.gz
STAR reference build
STAR aligner reference build on Pronghorn
### Copy templet
cp /data/gpfs/assoc/bch709-5/students/Course_materials/human/run.sh /data/gpfs/assoc/bch709-5/students/${USER}/human/ref/ref_build.sh
#open text editor
### PLEASE RENAME EMAIL AND JOB NAME
sed -i "s/16g/64g/g; s/\-\-cpus\-per\-task\=2/\-\-cpus\-per\-task\=4/g; s/\[NAME\]/ref_build/g; s/\[youremail\]/${USER}\@unr.edu\,${USER}\@nevada.unr.edu/g" /data/gpfs/assoc/bch709-5/students/${USER}/human/ref/ref_build.sh
nano ref_build.sh
# Add below command to ref_build.sh
STAR --runThreadN 4 --runMode genomeGenerate --genomeDir . --genomeFastaFiles GRCh38_latest_genomic.fna --sjdbGTFfile GRCh38_latest_genomic.gtf --sjdbOverhang 99 --genomeSAindexNbases 12
Submit job to HPC
#submit job
sbach ref_build.sh
#check job
squeue -u ${USER}
FASTQ file
cd /data/gpfs/assoc/bch709-5/students/${USER}/human/
### Link file (without copy)
ln -s /data/gpfs/assoc/bch709-5/students/Course_materials/human/fastq/* /data/gpfs/assoc/bch709-5/students/${USER}/human/fastq
ls /data/gpfs/assoc/bch709-5/students/${USER}/human/fastq
Create file list
cd /data/gpfs/assoc/bch709-5/students/${USER}/human/fastq
ls -1 *.gz
ls -1 *.gz | sed 's/_R.\.fastq\.gz//g'
ls -1 *.gz | sed 's/_R.\.fastq\.gz//g' | sort -u
ls -1 *.gz | sed 's/_R.\.fastq\.gz//g' | sort -u > /data/gpfs/assoc/bch709-5/students/${USER}/human/filelist
cat /data/gpfs/assoc/bch709-5/students/${USER}/human/filelist
Regular expression
https://regex101.com/
Trim reads
Prepare templet
cp /data/gpfs/assoc/bch709-5/students/Course_materials/human/run.sh /data/gpfs/assoc/bch709-5/students/${USER}/human/fastq/trim.sh
sed -i "s/16g/64g/g; s/\-\-cpus\-per\-task\=2/\-\-cpus\-per\-task\=4/g; s/\[NAME\]/Trim/g; s/\[youremail\]/${USER}\@unr.edu\,${USER}\@nevada.unr.edu/g" /data/gpfs/assoc/bch709-5/students/${USER}/human/fastq/trim.sh
Edit templet
nano /data/gpfs/assoc/bch709-5/students/${USER}/human/fastq/trim.sh
Batch submission
# Check file list
cat ../filelist
nano trim.sh
# Loop file list
### Add Forward read to variable
### Add reverse read from forward read name substitution
### add file name from variable to trim-galore
### merge trim-galore command and trim.sh
### add trim-galore command and trim.sh to new file
for i in `cat ../filelist`
do
read1=${i}_R1.fastq.gz
read2=${read1//_R1.fastq.gz/_R2.fastq.gz}
echo "trim_galore --paired --three_prime_clip_R1 5 --three_prime_clip_R2 5 --cores 2 --max_n 40 --fastqc --gzip -o /data/gpfs/assoc/bch709-5/students/${USER}/human/trim $read1 $read2" | cat trim.sh - > ${i}_trim.sh
echo "$read1 $read2 trim file has been created."
done
Batch submission
ls -1 *_trim.sh
### Loop *.sh submission
for i in `ls -1 *_trim.sh`
do
sbatch $i
done
Check submission
squeue -u ${USER}
RNA-Seq Alignment
#### Move to trim folder
cd /data/gpfs/assoc/bch709-5/students/${USER}/human/trim
#### Copy templet
cp /data/gpfs/assoc/bch709-5/students/Course_materials/human/run.sh /data/gpfs/assoc/bch709-5/students/${USER}/human/trim/mapping.sh
sed -i "s/16g/64g/g; s/\-\-cpus\-per\-task\=2/\-\-cpus\-per\-task\=4/g; s/\[NAME\]/Mapping/g; s/\[youremail\]/${USER}\@unr.edu\,${USER}\@nevada.unr.edu/g" /data/gpfs/assoc/bch709-5/students/${USER}/human/trim/mapping.sh
#### Edit templet
nano mapping.sh
Check output
ls -algh /data/gpfs/assoc/bch709-5/students/${USER}/human/trim
Output example
[FILENAME]_R1_val_1.fq.gz [FILENAME]_R2_val_2.fq.gz
STAR RNA-Seq alignment
STAR --runMode alignReads --runThreadN 4 --outFilterMultimapNmax 100 --alignIntronMin 25 --alignIntronMax 50000 --quantMode TranscriptomeSAM GeneCounts --genomeDir /data/gpfs/assoc/bch709-5/students/${USER}/human/ref --readFilesCommand gunzip -c --readFilesIn /data/gpfs/assoc/bch709-5/students/${USER}/human/trim/[FILENAME]_R1_val_1.fq.gz /data/gpfs/assoc/bch709-5/students/${USER}/human/trim/[FILENAME]_R2_val_2.fq.gz --outSAMtype BAM SortedByCoordinate --outFileNamePrefix /data/gpfs/assoc/bch709-5/students/${USER}/human/bam/[FILENAME].bam
STAR RNA-Seq alignment batch file
cd /data/gpfs/assoc/bch709-5/students/${USER}/human/trim
for i in `cat ../filelist`
do
read1=${i}_R1_val_1.fq.gz
read2=${read1//_R1_val_1.fq.gz/_R2_val_2.fq.gz}
echo $read1 $read2
echo "STAR --runMode alignReads --runThreadN 4 --outFilterMultimapNmax 100 --alignIntronMin 25 --alignIntronMax 50000 --genomeDir /data/gpfs/assoc/bch709-5/students/${USER}/human/ref --outSAMtype BAM SortedByCoordinate --readFilesCommand gunzip -c --readFilesIn /data/gpfs/assoc/bch709-5/students/${USER}/human/trim/${read1} /data/gpfs/assoc/bch709-5/students/${USER}/human/trim/${read2} --outFileNamePrefix /data/gpfs/assoc/bch709-5/students/${USER}/human/bam/${i}.bam" | cat mapping.sh - > ${i}_mapping.sh
done
Job submission dependency
squeue --noheader --format %i --user ${USER}
squeue --noheader --format %i --user ${USER} | tr '\n' ':'
Job submission dependency on Trim
jobid=$(squeue --noheader --format %i --user ${USER} | tr '\n' ':')1
for i in `ls -1 *_mapping.sh`
do
sbatch --dependency=afterany:${jobid} $i
done
FeatureCounts
Bioinformatics, Volume 30, Issue 7, 1 April 2014, Pages 923–930
featureCounts -o [output] -T [threads] -Q 1 -p -M -g gene_id -a [GTF] [BAMs]
FeatureCounts location
#### Move to trim folder
cd /data/gpfs/assoc/bch709-5/students/${USER}/human/bam
#### Copy templet
cp /data/gpfs/assoc/bch709-5/students/Course_materials/human/run.sh /data/gpfs/assoc/bch709-5/students/${USER}/human/bam/count.sh
sed -i "s/16g/64g/g; s/\-\-cpus\-per\-task\=2/\-\-cpus\-per\-task\=4/g; s/\[NAME\]/Count/g; s/\[youremail\]/${USER}\@unr.edu\,${USER}\@nevada.unr.edu/g" /data/gpfs/assoc/bch709-5/students/${USER}/human/bam/count.sh
FeatureCounts command to count.sh
LOOP example
cd /data/gpfs/assoc/bch709-5/students/${USER}/human/bam
ls -1 *.bam
for i in `cat /data/gpfs/assoc/bch709-5/students/${USER}/human/filelist`
do
echo ${i}.bamAligned.sortedByCoord.out.bam | tr '\n' ' '
done
FeatureCount
echo "featureCounts -o /data/gpfs/assoc/bch709-5/students/${USER}//mouse/readcount/featucount -T 4 -Q 1 -p -M -g gene_id -a /data/gpfs/assoc/bch709-5/students/${USER}/human/ref/GRCh38_latest_genomic.gtf $(for i in `cat /data/gpfs/assoc/bch709-5/students/${USER}/human/filelist`; do echo ${i}.bamAligned.sortedByCoord.out.bam| tr '\n' ' ';done)" >> count.sh
Job submission dependency
squeue --noheader --format %i --user ${USER}
squeue --noheader --format %i --user ${USER} | tr '\n' ':'
Job submission dependency on Align
cd /data/gpfs/assoc/bch709-5/students/${USER}/human/bam
jobid=$(squeue --noheader --format %i --user ${USER} | tr '\n' ':')1
sbatch --dependency=afterany:${jobid} count.sh
####################################
Slurm